Note: 10x Genomics does not provide support for community-developed tools and makes no guarantees regarding their function or performance. Please contact tool developers with any questions. If you have feedback about Analysis Guides, please email analysis-guides@10xgenomics.com.
Single-cell RNA-seq (scRNA-seq) has been widely used to characterize the transcriptomics landscapes in individual cells, facilitating cell atlas profiling, new cell type identification, and disease understanding. However, it is well known that different scRNA-seq datasets generated from different experiments suffer from batch effects. The research community has made many efforts to minimize the batch variations (Stuart et al. 2019, Haghverdi et al. 2018, Korsunsky et al. 2019, Polański et al. 2020). Despite the availability of in-silico batch correction algorithms for scRNA-seq data, it is good practice to design experiments that minimize batch differences from the very beginning. Sample multiplexing has been attracting more and more research attention to address batch effects, and since 2017, numerous multiplexing methods have been developed successively. Sample multiplexing has the added benefits of facilitating multiplet identification, lowering experiment costs, and making large-scale sample operations practical. In this article, we introduce main sample multiplexing methods along with the bioinformatics tools for sample demultiplexing.
The two main methods adopted for sample multiplexing are barcode-based and single nucleotide polymorphism (SNP)-based.
Barcode-based multiplexing
In barcode-based multiplexing, samples are labeled with unique DNA barcodes using two main strategies:
- Antibody tagged with DNA barcode that targets the cell surface protein or nucleus pore complex, i.e., cell hashing (Stoeckius et al. 2018) or nucleus hashing (Gaublomme et al. 2019);
- Lipid/cholesterol-modified oligonucleotide (LMO/CMO) tags cell membrane - MULTI-seq (McGinnis et al. 2019) and 10x Genomics CellPlex;
Additionally, a probe-based RNA detection assay called Single Cell Gene Expression Flex has been developed to allow the RNA profiling in a sample-multiplexing fashion. In this assay, pre-designed RNA-targeting probes containing probe barcodes are used for sample labeling.
In addition to direct sample labeling, sample barcodes can be indirectly introduced into individual samples by the viral integration of DNA barcodes into the genome. Once incorporated, these genetic barcodes are transcribed and can be captured in the transcriptomic data (CellTag Indexing - Guo et al. 2019).
SNP-based multiplexing
In SNP-based multiplexing, multiplexed samples are distinguished based on their natural genetic landscapes (i.e. SNPs). Genetically distinct samples can be pooled together for scRNAseq experiments and then cells for individual samples can be separated by harnessing genetic differences. The SNP based method of multiplexing requires fewer additional processing steps and is well-integrated into the current scRNAseq workflow. However, such an approach cannot be applied to multiplex samples sharing the same genetic signatures, such as samples from the same individuals at different developmental/lineage/experimental stages. Besides, additional genotyping data is needed in order to assign cells back to the samples of origin.
Both barcode and SNP-based multiplexing approaches are compatible with the 10x Chromium Next GEM platform, although not all of them are fully supported by 10x Genomics.
With the growing popularity of sample multiplexing, many demultiplexing tools are being developed in the community.
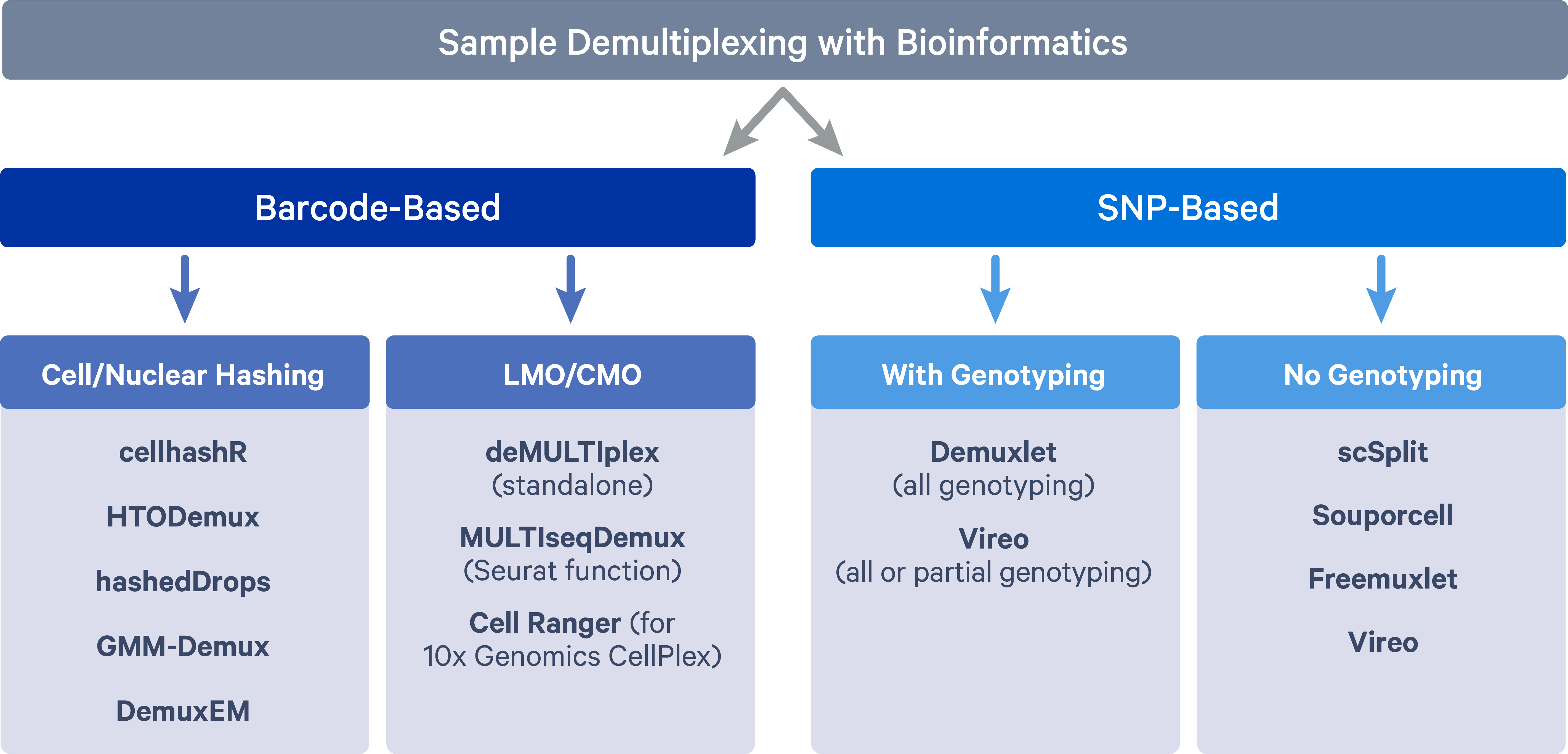
Barcode-based tools
Cell/Nuclear hashing:
1. cellhashR Bimodal Flexible Fitting (BFF) algorithm in cellhashR is based on a single assumption: a sample barcode count distribution is bimodal. Two algorithms are implemented to identify negatives (background), singlets, and doublets, BFFraw and BFFcluster. In the first step, BFF determines the count threshold for each sample barcode. After threshold determination, users can choose either BFFraw or BFFcluster mode. BFFraw directly uses the sample barcode thresholds to identify negatives, singlets, and doublets. BFFcluster takes the count thresholds to split positive and negative counts for subsequent normalization by Bimodal Quantile Normalization. Then the distributions of highest and second-highest counts are utilized to determine thresholds for negatives, singlets, and doublets.
- Publication: BFF and cellhashR: analysis tools for accurate demultiplexing of cell hashing data
- Source codes: cellhashR
- Tutorial: CellhashR general usage tutorial
2. HTODemux K-medoid clustering based on normalized hashtag oligo (HTO) counts is used to separate cells into K+1 groups (K is the number of samples). For each HTO, the cluster with the lowest average count is used as negative and this negative cluster is fitted to a negative binomial distribution. The 0.99 quantile (by default) of the distribution is set as a threshold to determine the positive or negative for this HTO.
- Source codes: Seurat
- Tutorial: Demultiplexing with hashtag oligos (HTOs)
3. hashedDrops The HTO abundances are adjusted by removing ambient contamination. For each barcode, the adjusted HTO count is added to a pseudo-count (average of the scaled ambient) to avoid zero after subtracting the ambient noise and then used to calculate two-fold change (FC) values: 1) the most abundant HTO vs the second-most abundant HTO, and 2) the second-most abundant HTO vs the ambient contamination. The first FC measures the confidence of assignment to a single sample and the second FC indicates the confidence of assignment to doublet.
- Source codes: DropletUtils
- Tutorial: Demultiplexing hashed libraries using hashedDrops
4. GMM-Demux Gaussian mixture model (GMM)-Demux fits the HTO UMI counts to the distribution of an aggregated two-Gaussian distribution mixed model. The Gaussian distribution with the smaller mean represents cell-free barcodes, while the other with the larger mean accounts for cell-containing barcodes. Based on the model, GMM-Demux computes the probabilities of barcodes to be single-sample droplets or multi-sample multiplets. Besides, GMM-Demux can estimate the percentages of single-sample multiplets (multiple cells from the same sample in a GEM) and singlets (only one cell in a GEM) in single-sample droplets. GMM-Demux is also able to identify whether a homogeneous GEM cluster is pure-type (only one cell type in a GEM), phony-type (multiple cell types in a GEM), or mixture (pure-type and phony-type).
- Publication: GMM-Demux: sample demultiplexing, multiplet detection, experiment planning, and novel cell-type verification in single cell sequencing
- Source codes: GMM-Demux
- Tutorial: GMM-Demux general usage tutorial
5. DemuxEM DemuxEM estimates the background hashtag counts based on empty barcodes. An expectation–maximization (EM) algorithm is used to estimate the fraction of hashtag counts from the background in each barcode. After deducting background counts and getting signal hashtag counts, if the signal hashtag count < 10, the barcode is unassigned; if the signal hashtag count >= 10, the number of tag types (samples) with >= 10% signal hashtag counts is counted. If such a number is 1, the barcode is a singlet; otherwise, it is a multiplet.
- Publication: Nuclei multiplexing with barcoded antibodies for single-nucleus genomics
- Source codes: demuxEM
- Tutorial: DemuxEM general usage tutorial
LMO/CMO:
1. deMULTIplex (standalone) / MULTIseqDemux (Seurat function) for MULTI-seq The algorithm uses the local maxima in a fitted count distribution of sample barcodes (Gaussian-kernel density estimation) to define signals from positive cells and background cells. Based on the defined local maxima, the algorithm finds a barcode-specific threshold across the inter-maxima quantile so that the proportion of singlets can be maximized. Then barcode-specific threshold is used to determine negatives, singlets, and doublets. In the end, negative barcodes are removed and the steps are repeated until no negative barcodes are left.
- Publication: MULTI-seq: sample multiplexing for single-cell RNA sequencing using lipid-tagged indices
- Source codes (deMULTIplex): MULTI-seq
- Tutorial (deMULTIplex): deMULTIplex general usage tutorial
- Tutorial (MULTIseqDemux): Demultiplex samples based on classification method from MULTI-seq
2. Cell Ranger for 10x Genomics CellPlex (CMO) Only the cell-containing barcodes filtered based on gene expression UMIs are used for CMO tag assignment. Tag counts are modeled into a normal distribution for each CMO type. Background mean, signal mean, and variance are estimated based on the observed tag counts. For each barcode, Cell Ranger calculates the likelihood of different states, including background, states for different single CMO, and multiplets. If the probability of any particular state is greater than 0.9 (default, can be adjusted), that state is assigned to the barcode. If no state has >0.9 probability, the barcode remains unassigned and is filtered out.
- Software: Cell Ranger
- Tutorial: Cell Multiplexing with cellranger multi
Despite their categorization based on the initial assay types they were developed for, (i.e. cell/nuclear hashing and LMO/CMO), in theory, all these tools may be used for both assay types. Here is an example to show how HTODemux is used to demultiplex samples tagged with CMOs: Tag Assignment of 10x Genomics CellPlex Data Using Seurat’s HTODemux Function.
SNP-based tools
1. Demuxlet Based on the genotyping data for the multiplexed samples and piled scRNA-seq reads across the reported genetic variants, Demuxlet determines the most likely donor for each barcode.
- Publication: Multiplexed droplet single-cell RNA-sequencing using natural genetic variation
- Source codes: demuxlet
- Tutorial: Demuxlet general usage tutorial
2. Freemuxlet Freemuxlet is a genotyping-free version of Demuxlet, which only requires a list of common SNP loci from a reference, such as Genome 10k. Based on the piled scRNA-seq reads across the reference variant sites, barcodes are assigned to N sample clusters (N is the input sample number based on experiment design), in addition to doublet and ambiguous groups.
- Source codes: popscle
- Tutorial: Freemuxlet general usage tutorial
3. Souporcell Souporcell is a genotyping-free tool to perform SNP-based demultiplexing. It provides the pipeline to remap reads using minimap2, call variant candidates using freebayes (this step can be replaced with an available source of reliable common variants), and count alleles per cell using vartrix. Cells are clustered based on the number of reads supporting each allele of the variant. In the end, given the cluster allele counts after clustering, barcodes are assigned as singlets or doublets.
- Publication: Souporcell: robust clustering of single-cell RNA-seq data by genotype without reference genotypes
- Source codes: souporcell
- Tutorial: Souporcell general usage tutorial
4. Vireo Vireo is a SNP-based sample demultiplexing tool that can be used with no, partial, or complete genotyping data. Vireo recommends a companion tool called cellSNP to generate sparse genotype data for all cells based on input common variants. These common variants can be derived from the existing projects, such as Genome 10K. If common variants are not available, variant calling can be performed directly from scRNA-seq using cellSNP or freebayes. The obtained sparse genotype data for all cells allows the reconstruction of partial genotypic states for the multiplexed samples and then for sample demultiplexing.
- Publication: Vireo: Bayesian demultiplexing of pooled single-cell RNA-seq data without genotype reference
- Source codes: vireo
- Tutorial: Vireo general usage tutorial
5. scSplit scSplit is a genotyping-free demultiplexing tool. As the first step, data quality control and filtering are performed on the bam file. Based on the filtered bam file, SNVs are called using freebayes, and allele count matrices are subsequently built. The most informative sub-matrices are generated and the initial allele fraction model with cluster representatives is constructed. The allele fraction model is further optimized using the expectation-maximization algorithm, and cells are assigned to their respective sample clusters. Presence/Absence matrix of alternative alleles is generated from the cell assignments.
- Publication: Genotype-free demultiplexing of pooled single-cell RNA-seq
- Source codes: scSplit
- Tutorial: scSplit general usage tutorial
Required skills and resources
- Despite the availability of multiple genotyping-free tools for SNP-based sample demultiplexing, genotyping data is required to map cells back to original sample individuals.
- Sample demultiplexing tools require familiarity with R/python programming or Linux commands
Things to watch out for
- SNP-based sample multiplexing requires distinct genetic variant landscapes across samples. Hence, it cannot be used to multiplex samples obtained from the same individuals at different time points, developmental stages, or experimental conditions.
- For cell hashing or LMO/CMO, it is good practice to visualize the tag count distribution before applying any methods. Gaining some basic understanding of raw data can help fine-tune the algorithm, if needed.
Analysis Tip
For SNP-based sample demultiplexing, if genotyping data is available for partial samples, in addition to Vireo, users can choose to combine the demultiplexing results from Demuxlet and another genotyping-free tool, such as Freemuxlet.