Note: 10x Genomics does not provide support for community-developed tools and makes no guarantees regarding their function or performance. Please contact tool developers with any questions. If you have feedback about Analysis Guides, please email analysis-guides@10xgenomics.com.
Single cell and Visium gene expression data can be combined to elucidate spatiality in single cell data and improve resolution in Visium data.
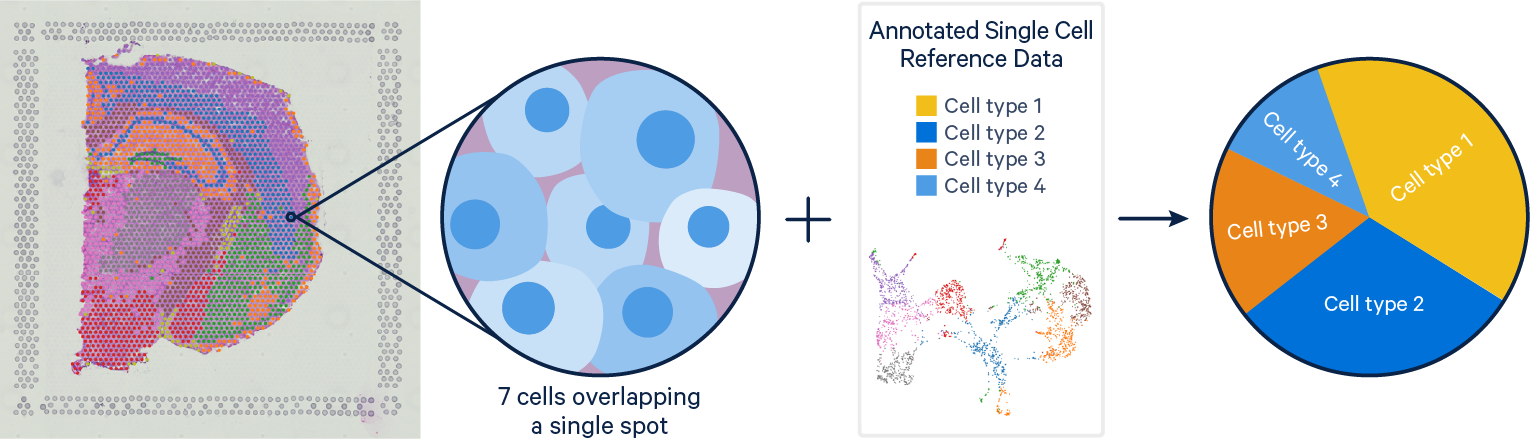
Problem: Single Cell RNAseq methods resolve gene expression at the single cell level, but lose the spatial context of the cells. Visium spatial gene expression maintains spatial information, but the resolution of each spot can cover is limited with each spot covering multiple cells (typically 1-10 cells).
Solution: With their complementary strengths these data types are a prime target for integration. The current computational tools for single cell and Visium integration fall on a continuum between deconvolution and mapping approaches. Deconvolution methods aim to identify the cell types and their relative proportions contributing to a spot, while mapping methods seek to assign the most likely dominant cell type to a spot.
For integration to work well, the single cell and Visium datasets need to be similar in their biology, but they do not necessarily need to be from the same sample. This allows the use of previously published and well characterized single cell datasets from large consortiums (eg, Human Cell Atlas) to be leveraged in analyzing Visium data. For examples of use cases, including cell-cell communication and rare identifying rare cell populations, and a more comprehensive analysis of deconvolution and mapping see the review cited below.
The selection of tools and algorithms below is not comprehensive. New and exciting tools, algorithms, and other resources continue to be released. We compiled this list based on a combination of factors including citations, quality of documentation, functionality/ease of use, and active support.
Spacexr/Robust cell-type decomposition (RCTD):
Deconvolution approach that uses a reference-based probabilistic model to resolve cell types from a single spot containing a mixture of cell types, infers the cell type proportions with a maximum-likelihood estimation, and projects them onto a spatial map of cell types.
- Publication: Cable, Dylan M., et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nature Biotechnology (2021)
- Tool: spacexr
- Tutorial: Applying CSIDE to Spatial Transcriptomics Data: Multiple regions in Visium with full mode RCTD
- Analysis Guide: Integrating 10x Visium and Chromium data with R
Seurat label transfer:
Mapping approach that can be used to “anchor” diverse datasets together, including different types of single cell data (transcriptomic, epigenomic, and proteomic) and single cell and spatial data.
- Publication: Stuart, Tim, et al. Comprehensive Integration of Single-Cell Data. Cell (2019)
- Tool: Seurat
- Tutorial: Analysis, visualization, and integration of spatial datasets with Seurat
Cell2location:
Deconvolution approach that can “incorporate prior information about the tissue to estimate absolute cell type abundance” as a Bayesian prior.
- Publication: Kleshchevnikov, Vitalii, et al. Cell2location maps fine-grained cell types in spatial transcriptomics. Nat Biotechnol (2022)
- Tool: Cell2location
- Tutorial: Mapping human lymph node cell types to 10X Visium
Tangram:
Mapping approach that takes advantage of graphics processing hardware (GPUs) for rapid run times.
- Publication: Biancalani, Tommaso, et al. Deep learning and alignment of spatially resolved single-cell transcriptomes with Tangram. Nat Methods (2021)
- Tool: squidpy
- Tutorial: Cell-type deconvolution using Tangram
STdeconvolve:
A reference free deconvolution approach that uses methods developed for natural language processing to identify cell types/states as “topics” from spot gene expression profiles. Though this is not technically integrating single cell and Visium data the objective is similar.
- Publication: Miller, Brendan, et al. Reference-free cell type deconvolution of multi-cellular pixel-resolution spatially resolved transcriptomics data. Nat Communications (2022)
- Tool: STdeconvolve
- Tutorial: Getting started with STdeconvolve