Note: 10x Genomics does not provide support for community-developed tools and makes no guarantees regarding their function or performance. Please contact tool developers with any questions. If you have feedback about Analysis Guides, please email analysis-guides@10xgenomics.com.
Batch effects come from technical variation across samples. This can often be prevented with good experimental design. When it cannot, there are computational approaches that can help.
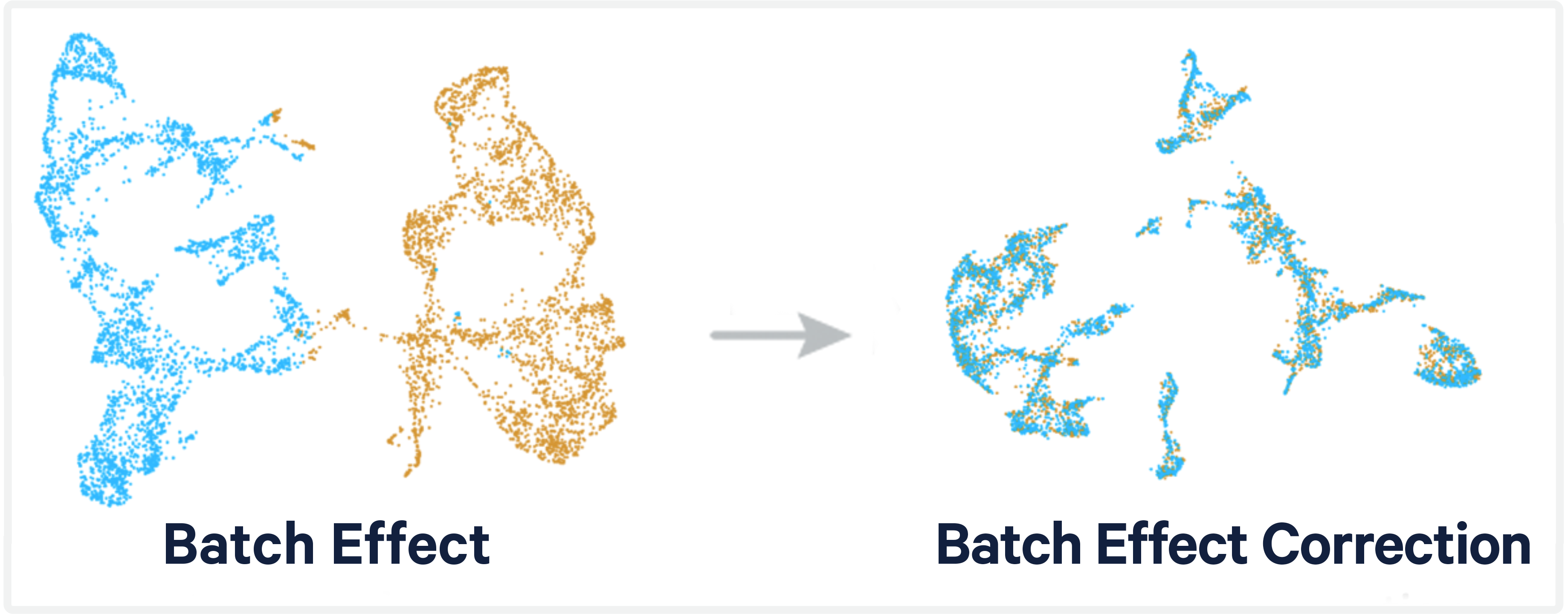
Problem: Variation in single-cell and spatial RNA sequencing data is known to be influenced by technical factors. In some cases, these technical factors may confound our ability to measure true biological variation between samples, making it more challenging to address the research question at hand.
Cause: These confounding factors include experimental biases and batch effects. Unavoidable systematic technical biases can include unequal amplification during PCR, cell lysis, reverse transcriptase enzyme efficiency, and stochastic molecular sampling during sequencing. By contrast, batch effects are technical, non-biological factors that also affect variation in the resulting data, but they occur in batches of samples. A “batch” refers to an individual group of samples that are processed differently relative to other samples in the experiment.
Solution: Technical factors that potentially lead to batch effects may be avoided with mitigation strategies in the lab and during sequencing. Examples of lab strategies include: sampling cells on the same day, using the same handling personnel, reagent lots, protocols, reducing PCR amplification bias, and generally using the same equipment. Sequencing strategies can include multiplexing libraries across flow cells. For example, if samples came from two patients, pooling libraries together and spreading them across flow cells can potentially spread out the flow cell-specific variation across samples.
Computational batch correction aims to remove technical variation from the data preventing this variation from confounding downstream analysis. There are several batch correction methods and tools that have implemented them.
The list below is not comprehensive. New and exciting tools, algorithms, and other resources continue to be released. We compiled this list based on a combination of factors including citations, quality of documentation, functionality/ease of use, and active support.
Harmony:
- Publication: Korsunsky, Ilya, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nature methods 16.12 (2019): 1289-1296.
- Tools: Harmony, harmonypy
- Tutorial: Harmony with Seurat V3, Integration of datasets using Harmony
- Analysis Guides: Correcting Batch Effects in Visium Data, Batch Effect Correction in Chromium Single Cell ATAC Data
Mutual Nearest Neighbors (MNN):
- Publications: Haghverdi, Laleh, et al. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nature biotechnology 36.5 (2018): 421-427.
- Tools: mmnpy, mnnCorrect, Batchelor
- Tutorials: Performing MNN correction, Running fastMNN on Seurat Objects
LIGER:
- Publication: Welch, Joshua D., et al. Single-cell multi-omic integration compares and contrasts features of brain cell identity. Cell 177.7 (2019): 1873-1887.
- Tool: LIGER
- Tutorial: Integrating Seurat objects using LIGER
Seurat Integration:
- Publication: Stuart, Tim, et al. Comprehensive integration of single-cell data. Cell 177 (2019): 1888–1902.
- Tool: Seurat
- Tutorial: Introduction to scRNA-seq integration