Behind the preprint: Combining single cell, spatial, and in situ for deeper biological understanding

In this blog post, we talk with two 10x Genomics scientists and co-authors of our recent preprint who analyzed serial sections from a single FFPE breast cancer tissue block using Chromium Single Cell, Visium Spatial, and the new Xenium In Situ platform in tandem. They reflect on the power of this multifaceted analysis and the importance of collaboration in this study.
Since this blog was released, our bioRxiv preprint was published in Nature Communications. Please take a look at the peer-reviewed, published article here!
“Great discoveries and improvements invariably involve the cooperation of many minds.”
~Alexander Graham Bell
Collaboration in biological research is a thing of beauty and necessity. The Human Cell Atlas, the International Cancer Genome Consortium, and the International Brain Laboratory are a few examples of brilliant researchers coming together to tackle some of human biology’s most complex mechanisms and diseases.
Combining multiple technologies that can approach these challenging biological questions from different angles is equally powerful.
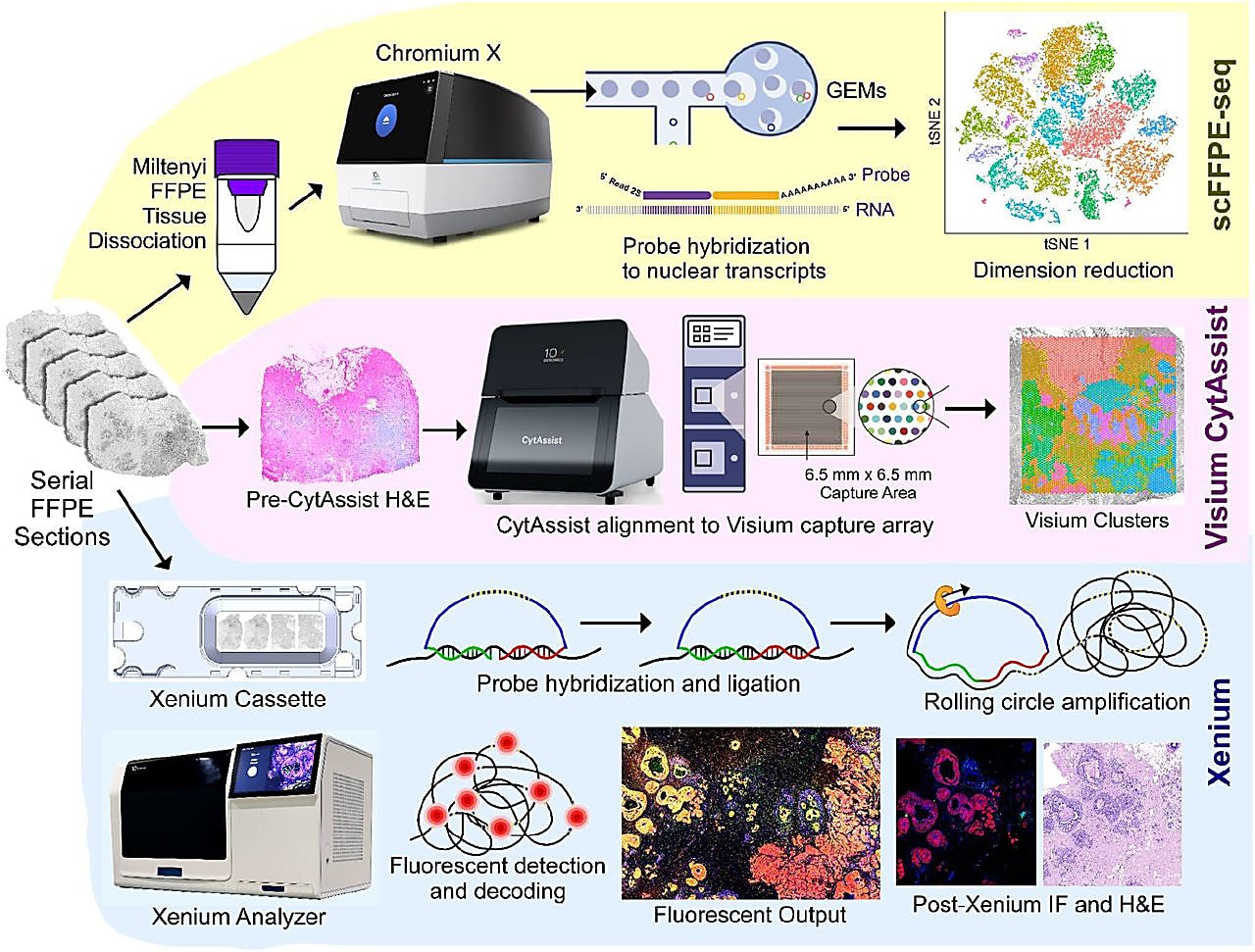
In a recent bioRxiv preprint, an interdisciplinary group of 10x Genomics scientists demonstrated this by applying Chromium single cell FFPE sequencing (scFFPE-seq), Visium Spatial Gene Expression for FFPE, and Xenium In Situ targeted gene expression to examine the tumor microenvironment of a single FFPE-preserved breast cancer tissue block (1). The synergy of these three groundbreaking genomic technologies, alongside the collaboration of several scientific groups at 10x Genomics, enabled a uniquely comprehensive investigation of tumor invasion in ductal carcinoma in situ (DCIS).

“I think this type of collaborative study is the future. One individual or technology alone cannot do anything. It’s when a group is collaborating with each other that great science happens,” said Morgane Rouault, PhD, a staff scientist at 10x Genomics and co-author on the new preprint.
Robert Shelansky, PhD, another of the preprint authors, echoed Dr. Rouault’s thoughts: “In this project there was a lot of going back and forth between myself and the cancer biologists. There was this one point in time where my colleague Amanda Janesick [the first author], found a subset of the tumor that had a distinct molecular signature. This prompted us to dig into that region across the spatial, scFFPE-seq, and in situ datasets to figure out what was going on. And, we were sitting there stunned at how quickly we could dig into that question. It was a great moment of collaboration not just for ourselves but with the technologies.”
One FFPE block, three technologies
“The preprint tells two stories,” reflects Dr. Rouault. “There is the biological story about molecular features associated with DCIS invasiveness that came out of the data, but there is also an important technology story.” The technological story is multifaceted, highlighting not only the strengths of each technology used but also how these technologies work together to build a more complete story.
It began with leveraging the whole transcriptome profiling approaches of Chromium scFFPE-seq and Visium Spatial for FFPE. scFFPE-seq allowed for the discovery of cell clusters present in FFPE sections taken from the DCIS sample.
While knowledge of the cell clusters present in the sample is useful, it is also necessary to know where those cells reside to truly understand their function. That is why the team also performed spatial gene expression analysis on tissue sections adjacent to the FFPE sections analyzed for scFFPE-seq. Importantly, the cell types annotated in the single cell data could then be mapped onto the clusters found within the Visium Spatial data pinpointing the location of distinct tumor domains.
Within the DCIS sample analyzed, there were some areas where the precise spatial localization of some cells could not be determined because multiple cell types were too close together to distinguish from one another. To spatially resolve the cells within these mixed clusters, the team performed targeted in situ gene expression analysis of 313 genes with Xenium In Situ (achieved with the 280 gene Xenium Human Breast Panel customized with 33 add-on genes). Relevant cell-type-specific genes from the panel were then used to make a spatial plot of the major cell types within the sample.
Bringing together the pieces of the puzzle
The biological question in the preprint, what makes some DCIS become invasive, is not a new one. Even with our current understanding of DCIS, the most common form of breast cancer, a large degree of uncertainty exists when it comes to predicting which DCIS cases will become invasive—estimates suggest anywhere from 20% to 50% of untreated DCIS cases transform to an invasive cancer (2).
A key element of this study is not just the application of these three genomic technologies to a single DCIS sample but combining the results for a deeper understanding of the biology being examined.
“I think the most interesting observation [to me] is that only with this holistic view of the different modalities, kind of collectively, can we observe some of the insights that we did,” commented Dr. Shelansky. For example, scFFPE-seq data allowed the determination of differences in cell-type proportions and differentially expressed genes that existed within unique regions of interest, representing distinct tumor microenvironments when mapped onto the Xenium data.
The preprint also demonstrates how these technologies can complement more traditional tissue technologies. The section analyzed by Xenium was processed with traditional hematoxylin & eosin staining for pathologist annotation due to the non-destructive nature of the Xenium workflow, which makes post-Xenium processing possible. The pathologist verified that the distinct tumor domains identified by scFFPE-seq—and validated by Visium and Xenium spatial data—did exhibit unique morphologies or microenvironments.
In fact, the preprint also demonstrates how these technologies were able to add insights to the pathology assessment. “So as an example to that, we show in the last figure of the paper that, with Xenium, we detected a tiny triple positive [HER2+/ER+/PR+] region in this block that was pathologically annotated as HER2+/ER+. Following this observation in the Xenium data, we then went back to the Visium genome-wide dataset and picked out individual genes that were differentially expressed in that particular [triple-positive] tumor region,” remarked Dr. Shelansky.
“Sometimes in research it feels like there are many different worlds. We, at 10x, live in the transcriptomic and spatial technology world. But we all know that there are also the histology and the pathology worlds out there. They can feel completely different but really at one point you want to have the single cell, imaging, and pathology experts and technologies interact fully with each other in order to make great discoveries,” added Dr. Rouault. “I think the preprint puts this very nicely together. The fact that we're using these technologies, but are also going back to the H&E staining is very relevant for translational research and clinicians. It's like the modern meeting the old, but actually, every piece of this is meaningful.”
Additionally, Dr. Shelansky noted that on top of adding insights to the pathology, the examination of the triple-positive region is a great example of how Xenium and Visium technologies came together. “We really couldn't have made these insights about the triple-positive region without the combination of the two. If we just did Xenium, we would have known that there's something funny going on in that one region, but wouldn't have had the whole genome to really dissect the actual context and biology in that region. Visium allowed us to go back and do that discovery-based assessment.”
Reflections from the bench
What did it take to pull off such a comprehensive study?
“On the biological side, there were some pragmatic concerns. One was the quality of FFPE-treated RNA in the block. Another, was making sure that there [was] enough of the block to use for all the parts of the study,” commented Dr. Rouault.
“Experimentally, I think the hard part is actually thinking about what things you need, like thinking about what pieces of biology can be resolved, is the challenging part,” added Dr. Shelansky. “You could always take a sample, do an experiment on it, and see what you see. You can fish for interesting spatial biology. But I think there are a lot of things that we know we can look for but don’t yet fully understand [based on existing research], and if we're careful about how we design our experiments we can now use these technologies with more powerful resolution to resolve the remaining questions.”
Computationally, things weren’t necessarily as complex as you might expect. “So when I was designing how I would present the computational analysis in this paper, I was very cognizant of how intimidating dealing with multiple datasets could seem. And I chose to use as little computation sophistication as possible,” noted Dr. Shelansky. “So, for example, when we wanted to do differential expression of different DCIS regions, we just drew ROI around each of these different tumor regions and directly compared them to each other rather than the more computationally heavy nearest neighbors analysis approach. The direct comparison approach we took can be done almost by hand.”
Dr. Shelansky said that having access to software that simplifies spatial analysis was also key to the success of this study. “The major computational challenge with in situ data is viewing the data. There's lots and lots of image data involved. If you want to look at a particular gene on this data, you need to make some kind of rendering of transcripts. Xenium Explorer software solves this problem for you. You can explore the data with very simple tools within the software and having that capability at your fingertips is really empowering.”
Paving the way for new explorations
Transcriptomic and spatial technologies have increased our understanding of many complex biological processes and diseases. However, there are still a lot of gaps in our knowledge that require higher resolution than any one technology can provide on its own. It will be through the combination of multiple experts and technologies that these gaps will begin to be filled.
“I think it will completely change so many things. The single cell revolution allowed you to ask questions about cell types and molecular signature. Spatial technologies then made it possible for you to ask questions about the relationships between those different cell types. Now, with high-plexy, targeted in situ panels, you can ask these amazing questions in a high-throughput manner across tissues. There's so much new biology that's at our fingertips to understand these relationships even better,” remarked Dr. Shelansky.
Want to learn more about this study? Watch this on-demand webinar to hear Drs. Rouault and Shelansky discuss the technologies and findings from the preprint (now published in Nature Communications).
Read our next blog to learn how Xenium In Situ uncovered a small triple-positive region in the breast cancer sample used for this study.
References:
- Janesick A, et al. High resolution mapping of the breast cancer tumor microenvironment using integrated single cell, spatial and in situ analysis of FFPE tissue. bioRvx (2022). doi: 10.1101/2022.10.06.510405
- Lesurf R, et al. Molecular features of subtype-specific progression from ductal carcinoma in situ to invasive breast cancer. Cell Reports 16: 1166–1179. doi:10.1016/j.celrep.2016.06.051
*Editor’s note: This article was updated in February 2024 with relevant content additions and links.*