From rare cells to rich insights with PERFF-seq

The Innovator Blog Series celebrates research conducted by 10x Genomics customers who have demonstrated scientific ingenuity by adapting Chromium Single Cell or Visium Spatial sequencing-based assays. These innovations are distinguished by their originality and potential impact on scientific discovery, including providing access to novel analytes, advancing multiomic analysis techniques, and demonstrating critical applications to human health and disease research. These techniques are customer developed, meaning they are not officially supported by 10x Genomics.
This article features a summary of the PERFF-seq method as well as a Q&A with first author Tsion Abay.
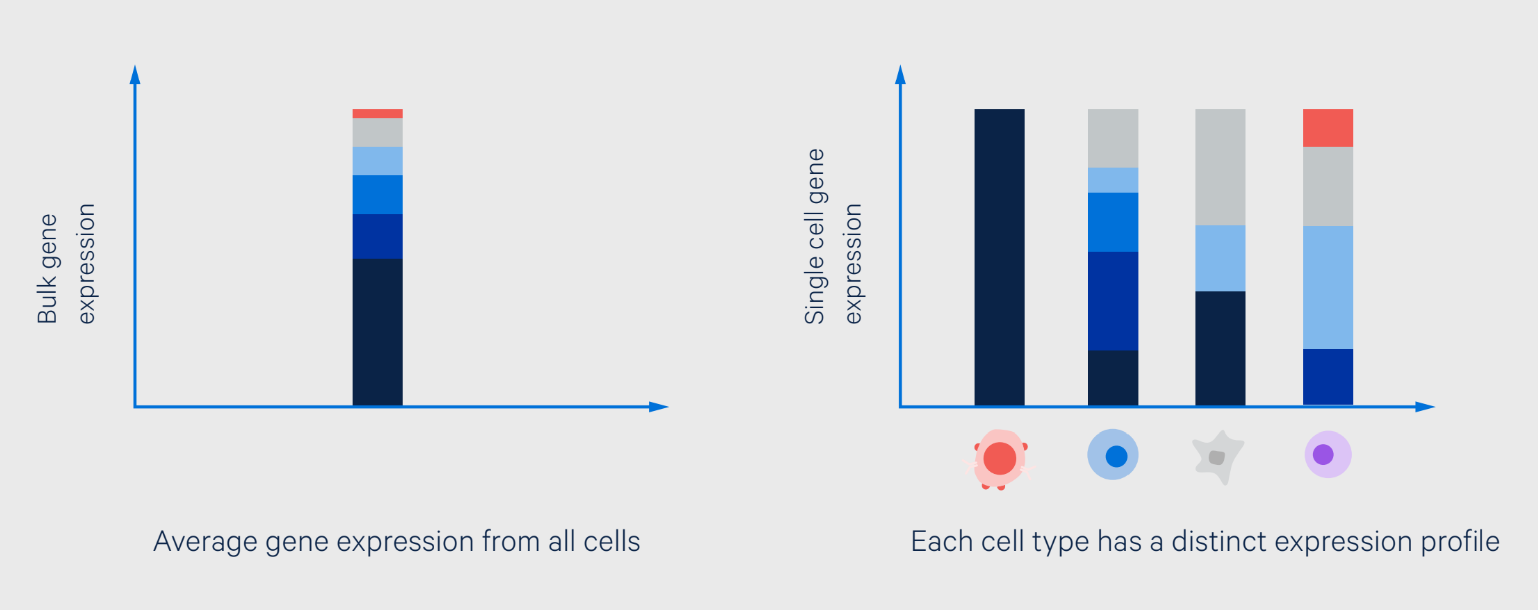
Biological heterogeneity appears at all levels—from the genetic- to the population-scale—and we now understand that it is the rule not the exception (1). The key challenge that researchers faced was identifying the differences, usually in the form of rare transcriptional signatures, behind the very real functional consequences they drive. That is, until single cell RNA-sequencing (scRNA-seq) allowed researchers to make direct measurements of transcriptional signatures in individual cells (Figure 1).
The identification of CFTR-expressing pulmonary ionocytes, which occur at a rate of 1 in 200 human lung epithelial cells, as potential mediators of cystic fibrosis pathology is just one of the well-known examples of pinpointing rare disease-associated cells (2, 3). Extraordinarily, scRNA-seq has been able to uncover even rarer cell types. For example, consider that scRNA-seq recently identified an incredibly rare population of CAR T cells that account for only ~1 in 10,000 cells in the infusion product, but contain high human herpesvirus 6 transcriptional activity, which results in reactivation of the HHV-6 virus (4).
Discovering rare cell types is just the first piece of the puzzle. Further characterization of these rare cells is needed to understand the regulatory networks that exist in these cells, identification of additional marker genes, and even examine heterogeneity within their populations. What is a researcher looking to dive deeper into these rare cell types to do?
While enrichment using fluorescence-activated cell sorting (FACS) might come to mind, in a new bioRxiv preprint, Abay et al. point out that it may not be feasible (3). The authors go on to list several challenges to performing FACS on rare cell populations, including:
- A lack of defined cell surface proteins specific to these rare cells that can be used for sorting
- Limited availability of high-quality antibodies for known cell surface proteins in these populations
- An inability to enrich or deplete using non-nuclear proteins when nuclei sequencing is required (often necessary when working with frozen or formalin-fixed fresh paraffin-embedded [FFPE] samples)
For Abay et al., the answer was to innovate a new method for enriching rare cells using RNA transcripts prior to performing scRNA-seq.
Meet PERFF-seq: Programmable Enrichment via RNA Flow-FISH by sequencing
PERFF-seq was originally a project spearheaded by Tsion Abay, Dr. Robert Stickels, and Dr. Caleb Lareau when they were trainees in Dr. Ansuman Satpathy's lab. Further development and applications of the technology happened when Dr. Lareau joined Memorial Sloan Kettering Cancer Center to start his own lab. There, he introduced collaborations with the Single-cell Analytics and Innovation Lab, which is headed by Dr. Ronan Chaligné, where Meril Takizawa is a senior research technician.
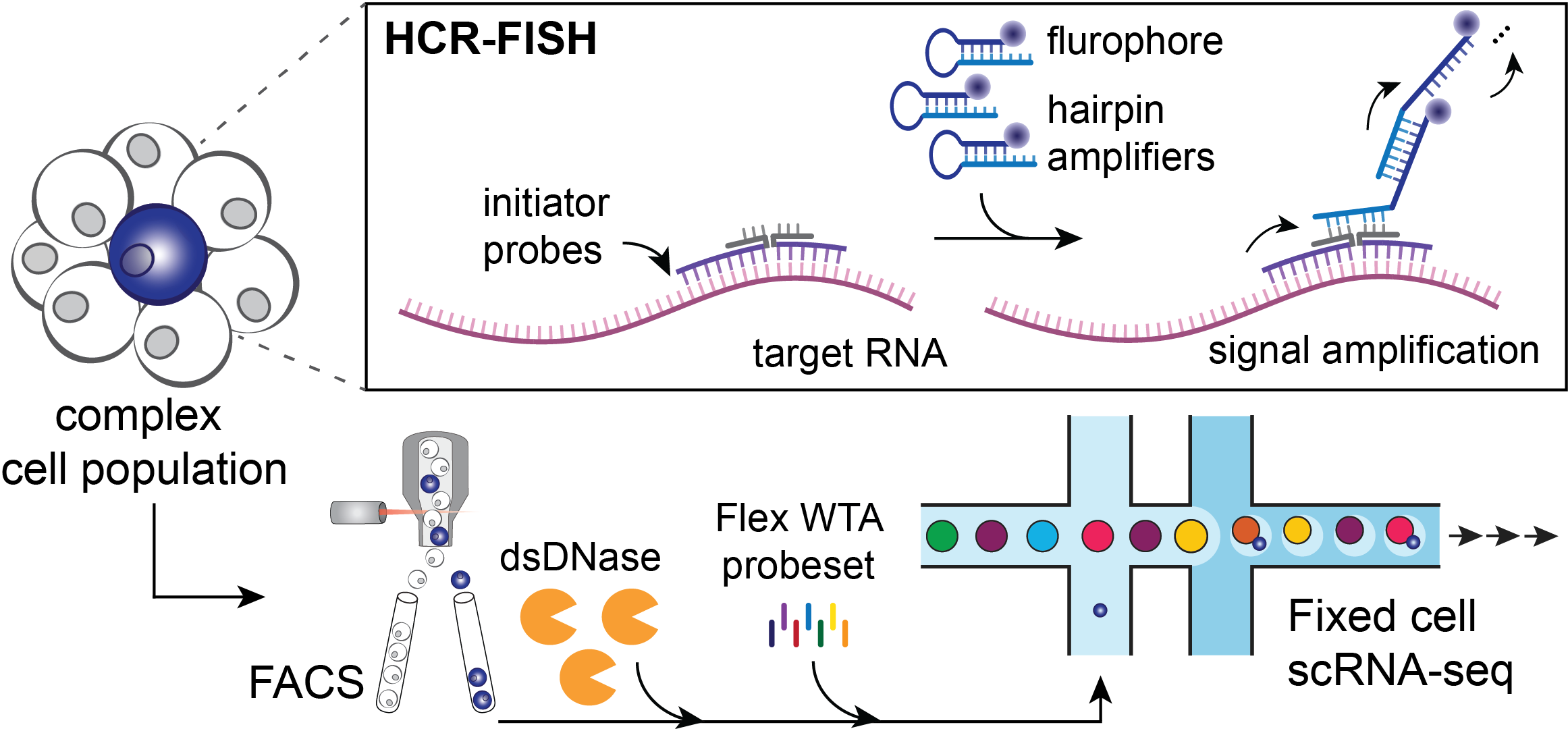
The PERFF-seq assay starts with processing a complex cell population (Figure 2) via hybridization chain reaction with fluorescence in situ hybridization (HCR-FISH). First, a pair of adjacent initiator probes bind to target RNA(s) that will be used for the sorting strategy. Next, hairpin amplifiers unzip and hybridize iteratively to generate an amplified fluorescent signal and enable FACS prior to single cell profiling with the Chromium Single Cell Gene Expression Flex kit.
While we could go on and on about our excitement for this method, we had the opportunity to connect with co-first author Tsion Abay and want to let you hear from her about the power of this new approach.

Tell me a little bit about the inception of this project? What inspired the development of the PERFF-seq method, and what were the specific gaps in existing technologies that you aimed to address?
When I joined Dr. Satpathy’s lab two years ago, there were many rare cell populations being discovered in the lab's genomics work. One example was cells in CAR T infusion products that are extremely rare at the beginning of culturing, about one in ten thousand, but then they eventually expand and infect other cells in culture. In parallel, there were reports of encephalitis in patients receiving CAR T as determined by positive HHV-6 tests. There was a link there that we were interested in, but it was hard to make any inferences beyond just the discovery of these cells, such as defining cell surface markers that could identify an HHV-g+ CAR T cell. In another project, the lab had identified rare antigen-presenting cells, and we had interest in better understanding the role of these cells in immune tolerance, but, again, these are extremely rare and difficult to study.
We were interested in being able to sort cells or isolate cells based on a unique transcriptional signature that we were seeing in these single cell readouts that we were doing. This technical limiation was the biggest gap that we were trying to fill in. Traditionally, people do protein marker–based sorting—so you can identify populations based on a surface protein expression that is unique to your cell type of interest, and then you can do a gene expression profile downstream. However, we really wanted to be able to profile cells based on the transcriptional signatures that we were seeing in single cell [analysis], and then be able to profile these cells further to make more meaningful inferences, such as heterogeneity within the rare populations.
Could you elaborate on the challenges you faced when developing the PERFF-seq technique, and how did you overcome them?
When we first started, the 10x [Chromium] Flex kit had come out, and we thought, “Oh, we can try to sort cells based on HCR-FISH—that has been shown to work really well in flow settings—and then be able to do 10x Flex because it enables this crosslinking that was previously not possible.”
When we did our first run, we only yielded ten genes detected per cell, which is really not good. So it was a kind of a “figure out where to start with troubleshooting this” process because both assays have really robust and rigorous workflows that have worked well before. However, putting them together was somehow causing this issue. This is where Bob’s, my co-first author on the paper [Dr. Robert Stickels], mentorship was super important because he has so much knowledge about FISH as an imaging method and has used it before for his own technology development.
We started decoupling the different parts of the HCR-FISH assay while maintaining the advantageous aspects that we wanted to keep, such as the high signal-to-noise ratio. After digging further, we found it was the HCR polymer that was causing a major issue, and we then found this enzymatic method that could allow us to get rid of this HCR polymer to enable the single cell Flex profiling downstream. After that, our vision for the method became more clear and we were able to expand to different sample and cell types of interest.
You touched on Chromium Flex being critical to the protocol. Can you talk about the attributes of Flex compared to other types of single cell RNA-seq you may have tried before?
I think we probably would not have thought to try this at all if the Flex kit hadn't come out because most other chemistries are reverse transcription–based. But, for this method, the crosslinking step is absolutely required for HCR-FISH so that we can access the RNA for binding probes. This Flex assay has really cool new chemistry that allowed for the crosslinking and enabled us to do this upstream.
Also, 10x protocols are extremely rigorous and robust. There's a lot of history of people building on top of these protocols, like Dr. Lareau with mtscATAC-seq and other groups with CITE-seq that enables your profile surface proteome.
One of the things that you touched on in the paper is the scalability of PERFF-seq. What implications do you think that has for large-scale genomic studies?
It’s really exciting that we are able to enrich based on an RNA marker of interest and enrich for really rare cell types that are otherwise difficult to study, and to do so in a non-laborious, [less] resource-intensive way.
Another implication is being able to do this kind of profiling in different sample contexts. Nuclei has traditionally been really difficult to work with because there aren’t many protein markers for sorting, and it can just be a difficult process to isolate and keep them alive long enough to do further profiling. But, with fixation and the additional enrichment based on RNA marker, I think that PERFF-seq it will be really enabling in studying rare populations from nuclei.
I think another setting this can be helpful for is studies using a transgenic mouse model or a cell line that traditionally would have to use genetic engineering to introduce fluorescent protein markers to be able to sort populations of interest. In these situations, where the researchers want to do gene expression profiling, it will be hugely beneficial to now have the option to sort out the RNAs based on their primary contexts and not based on an additional fluorescent protein marker that gets introduced.
Your study demonstrates the application of PERFF-seq in profiling immune cell subsets and brain tissue. Was there a particular finding that stood out to you as especially surprising or enlightening?
As I mentioned, nuclei have traditionally been really difficult to work with, and we were interested in this rare oligodendrocyte population (which make up ~2% of the total cells in the cerebellum) with a unique transcriptional marker that has been defined previously. So, we tried using these Mobp transcripts to sort out these cell types. We were really amazed by how well we were able to sort these nuclei, as evidenced in the single cell RNA-sequencing profiling, where we saw 98% enrichment in these cell types that have been previously difficult to isolate and enrich for.
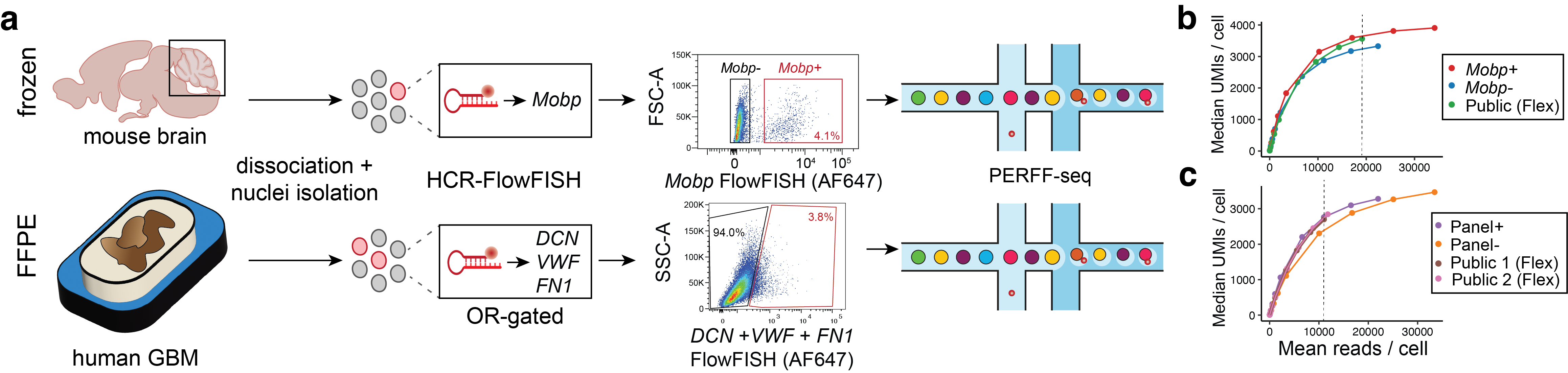
Are there any biological systems or disease systems for which you are really hoping PERFF-seq will be able to help find new and interesting insights?
It will be really exciting to see this used for any tissue samples that were previously difficult to profile due to limitations in previous single cell chemistries that will now be possible with the 10x Flex kit.
Additionally, I think the noncoding RNA space, which has also been previously very difficult to isolate and profile because you (by definition) don't have specific protein markers that you can find.
One final one, that is a little more specific, is studying host–pathogen interactions. With PERFF-seq, you can now sort cells based on a viral RNA or a bacterial RNA, and the RNA does not even have to be polyadenylated. Again, this will be possible, in part, because of the new Flex chemistry, but also the fact that you can sort out cells based on a viral, bacterial, or a parasitic transcript.
Do you have any advice for any researchers who are interested in adopting this method? How can they get started?
I would say that sorting based on RNA markers is different compared to sorting based on an antibody that's extremely specific to a protein. You kind of have to untrain your brain about how good you expect your signal-to-noise to look. If you have the right controls, you'll be able to distinguish between your true positive signal and something that could be background or nonspecific binding. Further, there may be brand new marker genes that could be super specific to your population that are not typically considered as they may be non-coding or intracellular, but would be valuable for the RNA-based sorting strategy of the PERFF-seq method.
You started working on this project while you were a research technician in the Satpathy lab. You’ve since moved on to start your graduate work at Harvard. Is single cell work going to continue to be a major focus of your research moving forward? What about PERFF-seq?
Definitely, yes. Both single cell and PERFF-seq will be helpful for the types of studies I’m interested in. I alluded to this, but I'm interested in host–pathogen interactions and understanding heterogeneity of infection in the immune context. I think it will be very enabling in looking at populations of cells infected with different pathogens to further understand the immune response, looking at heterogeneity in primary patient samples.
Also, in this context, oftentimes, researchers are really interested in dangerous pathogens that are Biosafety Level 4 or 3. With the Flex kit, enabling fixation allows the option of working with samples in less dangerous settings, which will be transformative for the kinds of questions I hope to pursue over the next few years.
Is there anything else that we didn't cover that you want to touch on?
Yes, so this work started when I was a research technician in the Satpathy lab. After I left and Dr. Lareau moved on to his role at Memorial Sloan Kettering Cancer Center, they were able to port this protocol over and it worked really well.
Dr. Lareau’s lab and the SAIL [Single-cell Analytics Innovation Lab] group have been instrumental in expanding this work and doing it in multiple different sample types. It's really awesome to see it being implemented at multiple institutions, and the method continues to produce high-quality data. We think this shows that it's a robust assay that can be broadly utilized by people in the single cell community.
We're also super excited about people using this assay, and that's why we have shared the protocol and data very openly. We are really hoping to see it being utilized broadly and want to see what further discoveries by PERFF-seq and our new understanding of how RNA FISH and single cell genomics can be utilized together.
References:
- Gough A, et al. Biologically relevant heterogeneity: Metrics and practical insights. SLAS Discov. 22: 213–237 (2017). doi: 10.1177/2472555216682725
- Montoro D, et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 560: 319–324 (2018). doi: 10.1038/s41586-018-0393-7
- Abay T, et al. Transcript-specific enrichment enables profiling rare cell states via scRNA-seq. bioRxiv. (2024). doi: 10.1101/2024.03.27.587039
- Lareau CA, et al. Latent human herpesvirus 6 is reactivated in CAR T cells. Nature. 623: 608–615 (2023). doi:10.1038/s41586-023-06704-2