Single cell DNA and RNA co-sequencing sheds light on the genotype–phenotype link in tumor heterogeneity

*Impact at a glance: A customer-developed method built on the Epi Multiome ATAC + Gene Expression assay, DEFND-seq, enables researchers to measure both DNA and RNA from individual nuclei. Its potential includes helping cancer researchers link driver mutations to distinct cellular phenotypes that cause tumor heterogeneity and reduce therapeutic efficacy—a notoriously difficult task that is now made possible by a method researchers can apply through a sample prep hack and Multiome reagents right out of the box.*
The Innovator Blog Series celebrates research conducted by 10x Genomics customers who have demonstrated scientific ingenuity by adapting Chromium Single Cell or Visium Spatial sequencing-based assays. These innovations are distinguished by their originality and potential impact on scientific discovery, such as providing access to novel analytes, advancing multiomic analysis techniques, and/or demonstrating critical applications to human health and disease research. These techniques are customer developed, meaning they are not officially supported by 10x Genomics.
A cellular view of heredity
It’s a question fundamental to all of biology: what is the relationship between an organism’s genome and its observable traits? It's a question that's captured the imagination of generations of scientists, including greats such as Darwin, Mendel, Johannsen, and others (1).
Now, focus that question on the single cell. What is the relationship between an individual cell’s genome, including the unique genetic variation it carries, and its transcriptome? How could defining that cellular genotype–phenotype relationship help to characterize the cell’s origin, identity, and function in a complex biological system composed of many heterogeneous cells (such as in the tumor microenvironment)?
A new method built on the Chromium Single Cell Multiome ATAC + Gene Expression assay could be the “ideal technology” to answer these kinds of questions. Called DEFND-seq in a recent bioRxiv pre-print (2), the approach leverages a sample prep method to deplete nuclei of nucleosomes—segments of DNA wound around histone proteins (3)—followed by single nuclei partitioning on the Chromium droplet microfluidic platform with Multiome ATAC + Gene Expression reagents to produce both mRNA and gDNA sequencing libraries from individual nuclei.
Commenting on the surprising accessibility of the method for other researchers, 10x Genomics VP of Product Strategy, Mike Lucero, said, “It used our product as-is—no modifications. It uses all the contents of our Multiome kit right out of the box. So that means customers can actually do it if they want to. They just need to read the paper.”
Salty sample prep opens up the genome for simultaneous single cell RNA- and DNA-seq
A sample prep hack enabled a team of researchers from Columbia University Irving Medical Center, led by Drs. Timothy Olsen and Pranay Talla, to use reagents from the Single Cell Multiome ATAC + Gene Expression assay to co-sequence genomic DNA and RNA in single nuclei. This sample prep approach allowed them to bypass the kit’s original use—measuring chromatin accessibility alongside RNA expression to better understand gene expression regulation.
Instead, the team treated the nuclei with lithium diiodosalicylate—a lithium salt and mild detergent used to extract histone proteins (2). This allowed them to disrupt chromatin packing in individual nuclei while maintaining the integrity of the nuclei for microfluidic partitioning and sequencing.
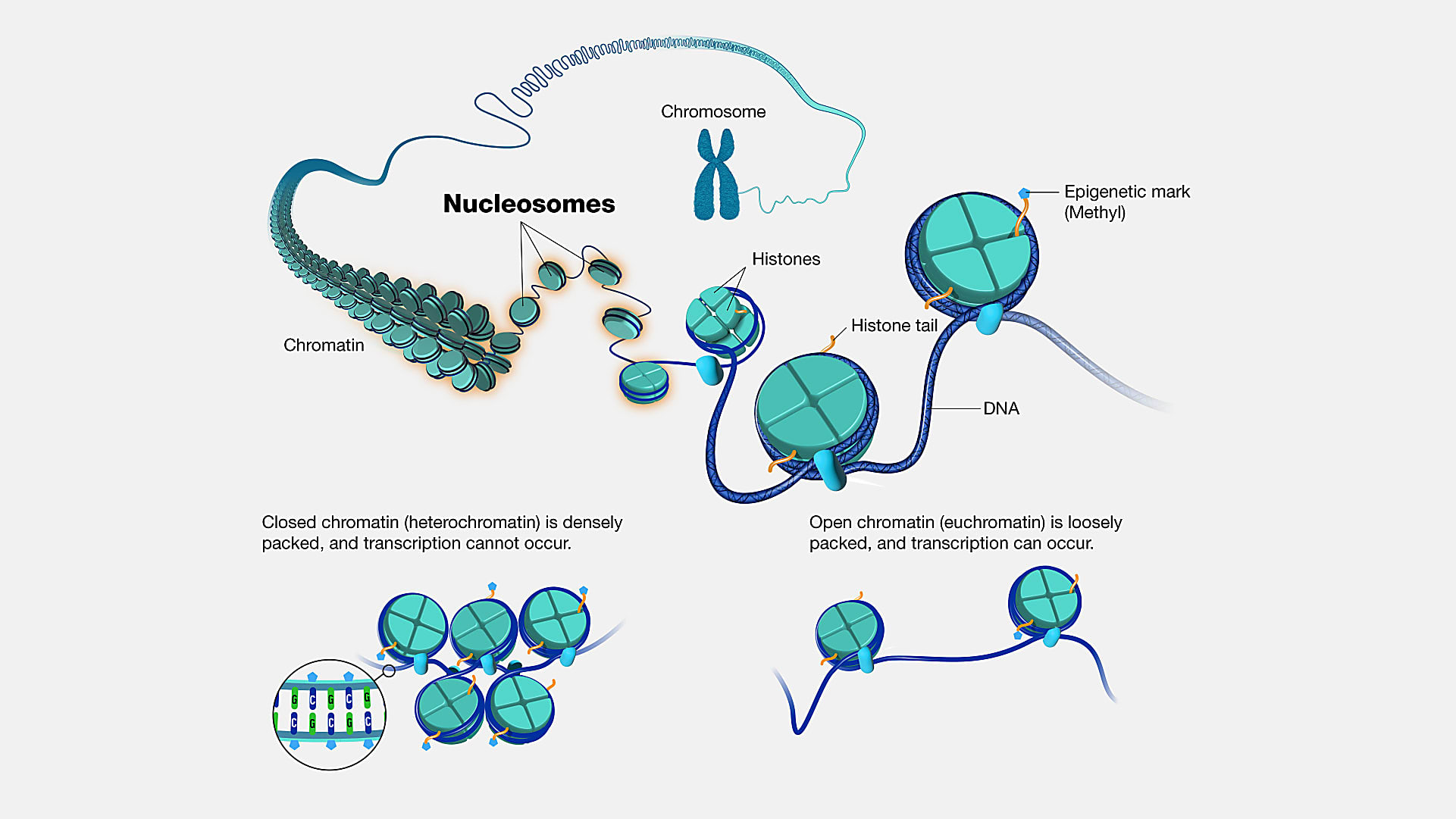
In the absence of histones, genomic DNA was extended and exposed. Following library preparation steps for the Single Cell Multiome ATAC + Gene Expression assay, nuclei were treated with reagents that included the enzyme transposase, which enters the nuclei and fragments DNA while adapter sequences are simultaneously added to the ends of the DNA fragments. Treated nuclei were simply loaded onto a microfluidic chip, which is run in a Chromium instrument to ultimately generate two sequencing libraries (mRNA and gDNA) for simultaneous analysis of the transcriptomes and genomes of individual nuclei (2).
10x Genomics’ Mike Lucero commented on the sensitivity of the method, saying, “It can do a really good job to produce a good genome, cell by cell. It may not be complete, but it’s the best thing I’ve seen come along to do it.”
Running on an established commercial system for single nuclei sequencing, DEFND-seq has an additional benefit: scalability. The authors noted that, while their validation studies used 4,400 nuclei per chip lane, they could easily scale up to 40,000 nuclei per sample with the 8-lane capacity of a standard 10x Genomics chip, and even potentially to over 100,000 nuclei if employing super-loading and demultiplexing (2).
“Sequencing has gotten less expensive. The advent of these very high-throughput sequencers with lower running costs—basically, for a $1,000 or less genome—means that people can really take advantage of that. Our product provides scale, so now you can afford to characterize thousands of cells, which is a meaningful number, whereas before it was out of reach for the typical lab, or most labs,” said Lucero.
The importance of the genotype–phenotype connection in glioblastoma and HPV-induced cancers
Of all other fields of research, cancer research may have the most to gain from employing a technique like DEFND-seq. “It’s going to meet a need for cancer researchers who want to uncover the heterogeneity of the cancer cells within a tumor, alongside a direct measurement of gene expression. So you can see the genotype–phenotype changes together,” said Lucero.
Certain cancer types may be perfectly suited for this method due to the role that genomic instability plays in driving cancer progression. For example, the DEFND-seq authors demonstrated the utility of the protocol in a cryopreserved glioblastoma surgical specimen.
Glioblastoma is notorious for its heterogeneity and phenotypic plasticity (2), which can wreak havoc on therapeutic efficacy. Moreover, while genome sequencing studies have characterized many key driver mutations in glioblastoma, like in TP53 (a gene that controls cell division and death (4)) or in the PI3K signaling pathway (which controls cell growth (4)), few are matched with an associated phenotypic subtype (2).
Yet with joint single cell RNA and DNA profiling, the Columbia University team was able to characterize six transformed cellular clusters—representing abnormal, neoplastic cells—with their associated genomic alterations. Specifically, transformed clusters carried a deletion in a region of Chromosome 10 that contains PTEN, a gene commonly associated with shorter survival in glioblastoma (2), and amplification of regions in Chromosome 2 and 12 that contain MYCN and CDK4, respectively, both of which are associated with tumorigenesis (2).
They also identified a missense point mutation in PREX1—part of the PI3K signaling pathway—in an astrocyte-like glioma cluster of 76 transformed cells that were enriched for PREX1 expression, thus linking a somatic single nucleotide variation event with a cellular phenotype (2).
Human papillomavirus (HPV)-induced cancers (frequently manifesting in the head and neck) represent another class of diseases for which it would be crucial to understand how genomic changes drive progression. Recent findings enabled by long-read sequencing show that cancer is likely caused by a process of viral DNA translocation within the host genome called heterocateny (5). In this model, HPV circular DNA replicates in the host nucleus, then inserts into the host cell’s genome; excision of viral DNA from the host genome will bring along host DNA segments that may contain oncogenes. Additional replication, insertion, and excision events can subsequently increase the copy number of that oncogene and cause genomic instability, which can promote cancer development (5).
The ability to trace complex genetic events such as these and link them to a clear cellular phenotype could be a game changer for researchers to deeply understand the basis of clonal evolution and define intratumoral heterogeneity, which could, in turn, improve therapeutic approaches and outcomes.
A new era of single cell molecular genetics
Though past models of genetic research have transformed our understanding of the relationship between the genome and an organism’s traits, they were also limited by what was observable with available technology. With the ability to perform joint single cell RNA and DNA sequencing, scientists can now observe the smallest units of biology (single cells), which coordinate together in complex organs to produce observable traits, and directly measure the genetic variation controlling those cells’ identities and functions, encoded in their transcriptomes.
Access to these once unobservable genotype–phenotype relationships has the potential to dramatically accelerate discovery of the development secrets that scientists have pursued for hundreds of years, and overcome knowledge gaps in cancer research blocking therapeutic advancement.
Learn more about joint single cell RNA and DNA sequencing in this preprint →
And explore the Chromium Single Cell Multiome ATAC + Gene Expression assay.
References:
- Taylor P and Lewontin R. "The Genotype/Phenotype Distinction," The Stanford Encyclopedia of Philosophy (Summer 2021 Edition), Edward N. Zalta (ed.). https://plato.stanford.edu/archives/sum2021/entries/genotype-phenotype/
- Olsen T, et al. Scalable co-sequencing of RNA and DNA from individual nuclei. bioRxiv (2023). https://doi.org/10.1101/2023.02.09.527940
- https://www.genome.gov/genetics-glossary/Nucleosome
- https://www.cancer.gov/publications/dictionaries/cancer-terms
- Akagi K, et al. Intratumoral heterogeneity and clonal evolution induced by HPV integration. Cancer Discov 13: 910–927 (2023). doi: 10.1158/2159-8290.CD-22-0900