Job submission mode is one of three primary ways of running Cell Ranger ARC. To learn about the other approaches, refer to the computing options page.
Cell Ranger ARC can be run in job submission mode, by treating a single node from the cluster like a local server. This leverages existing institutional hardware for pipeline analysis.
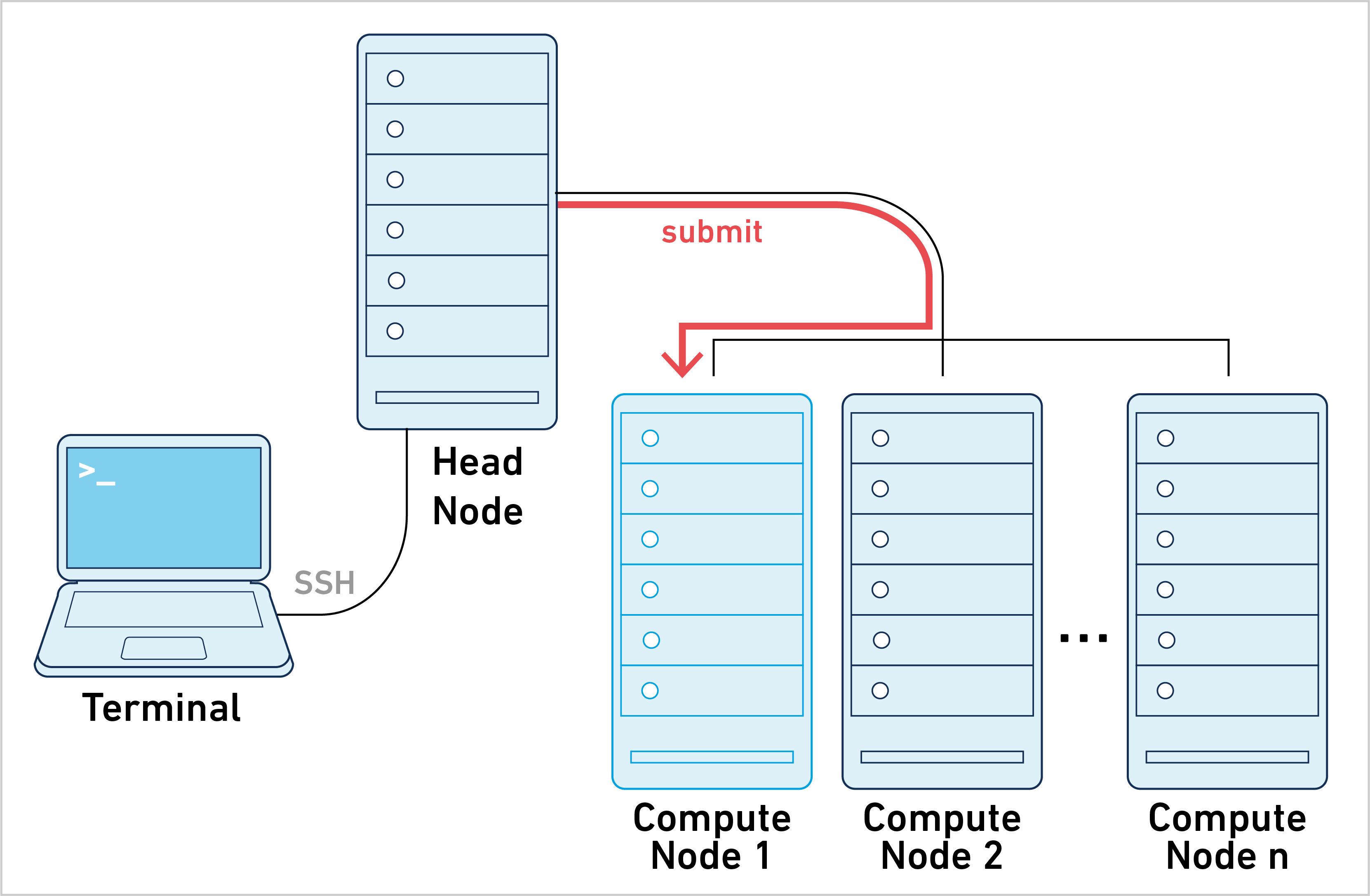
Running cellranger-arc commands in this mode is typically achieved by submitting a job script to the cluster. The process is as follows:
Step 1. Select a job scheduler tab below to see a sample job script.
Step 2. Copy the sample job script to a text editor and replace all the variables enclosed within braces (e.g. {JOB_NAME} or {STDOUT_LOG}) with literal names, paths, and values. The last line, {PIPELINE_CMD} , is replaced by a Cell Ranger ARC command, specifying --jobmode=local, --localcores={NUM_THREADS} and --localmem=0.9*{MEM_GB} to restrict CPU and memory use that is consistent with the directives in the job script. For example, if the job script requests 8 cores and 64 GB of memory from the cluster, then the Cell Ranger ARC command should include --jobmode=local --localcores=8 --localmem=57.
#!/usr/bin/env bash
#
# =============================================================================
# Setup Instructions
# =============================================================================
#
# 1. Substitute {PE_NAME} below with name of your cluster's shared-memory
# parallel environment. If your cluster does not have a parallel environment,
# delete this line. However, the job will run with only 1 thread.
#
# =============================================================================
# Job Script
# =============================================================================
#
#$ -N {JOB_NAME}
#$ -V
#$ -pe {PE_NAME} {NUM_THREADS}
#$ -cwd
#$ -l mem_free={MEM_GB}G
#$ -o {STDOUT_LOG}
#$ -e {STDERR_LOG}
#$ -S "/usr/bin/env bash"
{PIPELINE_CMD}
#!/usr/bin/env bash
#
# =============================================================================
# Job Script
# =============================================================================
#
#BSUB -J {JOB_NAME}
#BSUB -n {NUM_THREADS}
#BSUB -o {STDOUT_LOG}
#BSUB -e {STDERR_LOG}
#BSUB -R "rusage[mem={MEM_MB}]"
#BSUB -R span[hosts=1]
{PIPELINE_CMD}
#!/usr/bin/env bash
#
# =============================================================================
# Job Script
# =============================================================================
#
#SBATCH -J {JOB_NAME}
#SBATCH --export=ALL
#SBATCH --nodes=1 --ntasks-per-node={NUM_THREADS}
#SBATCH --signal=2
#SBATCH --no-requeue
### Alternatively: --ntasks=1 --cpus-per-task={NUM_THREADS}
### Consult with your cluster administrators to find the combination that
### works best for single-node, multi-threaded applications on your system.
#SBATCH --mem={MEM_GB}G
#SBATCH -o {STDOUT_LOG}
#SBATCH -e {STDERR_LOG}
{PIPELINE_CMD}
#!/usr/bin/env bash
#
# =============================================================================
# Job Script
# =============================================================================
#
#SBATCH -J {JOB_NAME}
#SBATCH --export=ALL
#SBATCH --nodes=1 --ntasks-per-node={NUM_THREADS}
#SBATCH --signal=2
#SBATCH --no-requeue
### Alternatively: --ntasks=1 --cpus-per-task={NUM_THREADS}
### Consult with your cluster administrators to find the combination that
### works best for single-node, multi-threaded applications on your system.
#SBATCH --mem={MEM_GB}G
#SBATCH -o {STDOUT_LOG}
#SBATCH -e {STDERR_LOG}
{PIPELINE_CMD}
Step 3. If this job script is saved as runJob.bash, then the following command submits the job to the cluster:
sbatch runJob.bash