Cell Ranger relies on a specific multi config CSV structure for proper execution. Failure to comply with the required format (e.g., column headers, delimiters) will lead to parsing errors.
Ensure that the multi config is saved in CSV format with the CSV extension.
This page explains how to analyze libraries multiplexed using three different methods with the cellranger multi pipeline. The available sample multiplexing techniques include:
Most common library combinations are described here. If your specific library combination is not shown and you need assistance, please contact 10x Genomics Support at support@10xgenomics.com
For general information on setting up and running the multi pipeline, visit the Cell Ranger multi pipeline page. Go to the Cell Ranger Multi Config CSV page for a complete list of options for each section.
To generate a multi config CSV template, run cellranger multi-template and see the usage instructions here.
Examples of multi config CSVs for the most common library combinations are provided here. If your specific library combination is not listed and you need assistance, please contact 10x Genomics Support at support@10xgenomics.com.
The GEM-X v4 4-plex assay provides a scalable microfluidic platform for on-chip multiplexing (OCM) of up to eight samples (two sets of up to four samples each). It enables the assessment of Gene Expression, Antibody Capture, V(D)J profiling, and CRISPR Guide Capture per sample, for both Universal 3' and 5' assays.
Example multi config CSVs for common library combinations with on-chip multiplexing are listed below. The [samples] section is still required if you perform a singleplex run on an OCM chip.
The samples section requires the ocm_barcode_ids field for demultiplexing.
For details, visit the multi config CSV documentation.
A complete list of input files required to process OCM libraries is available in the multi section of the List of inputs page.
If any lanes in a set contain mock sample (1X PBS with Master Mix), exclude those lanes from the [samples] section in the multi config CSV file.
In this example, 3' Gene Expression libraries were created from four samples that were multiplexed on-chip. Technical replicates are identified by different OCM barcodes (for 3', OB1 = blue, OB2 = red, OB3 = yellow, and OB4 = green).
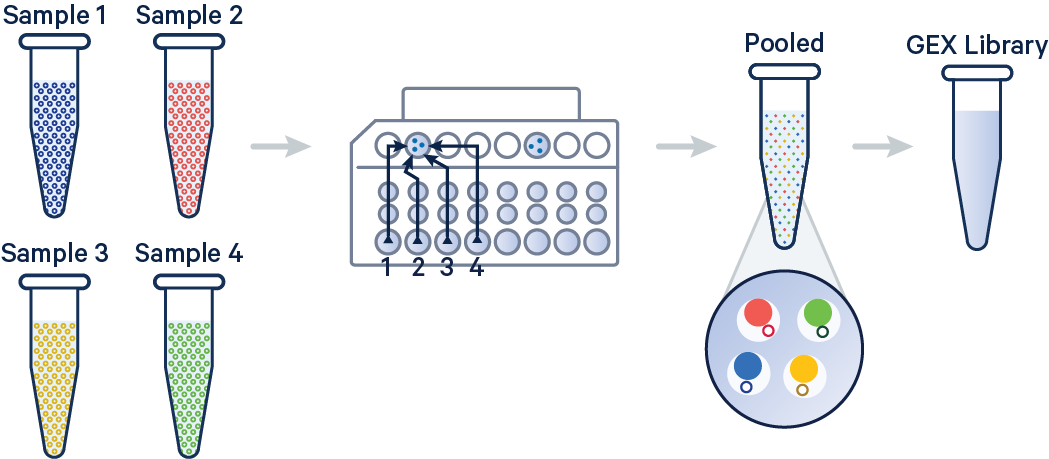
[gene-expression]
reference,/path/to/transcriptome
create-bam,false
[libraries]
fastq_id,fastqs,lanes,feature_types,
gex1,/path/to/gex_fastqs,Gene Expression
[samples]
sample_id,ocm_barcode_ids
sample1,OB1
sample2,OB2
sample3,OB3
sample4,OB4
In this example, 3' Gene Expression and Antibody Capture libraries were prepared from a single biological sample, which was divided across four GEM wells (or four OCM Barcodes) to generate technical replicates. Each library (Gene Expression and Antibody Capture) is linked to its corresponding FASTQ files. Technical replicates are identified by different OCM barcodes (for 3', OB1 = blue, OB2 = red, OB3 = yellow, and OB4 = green).
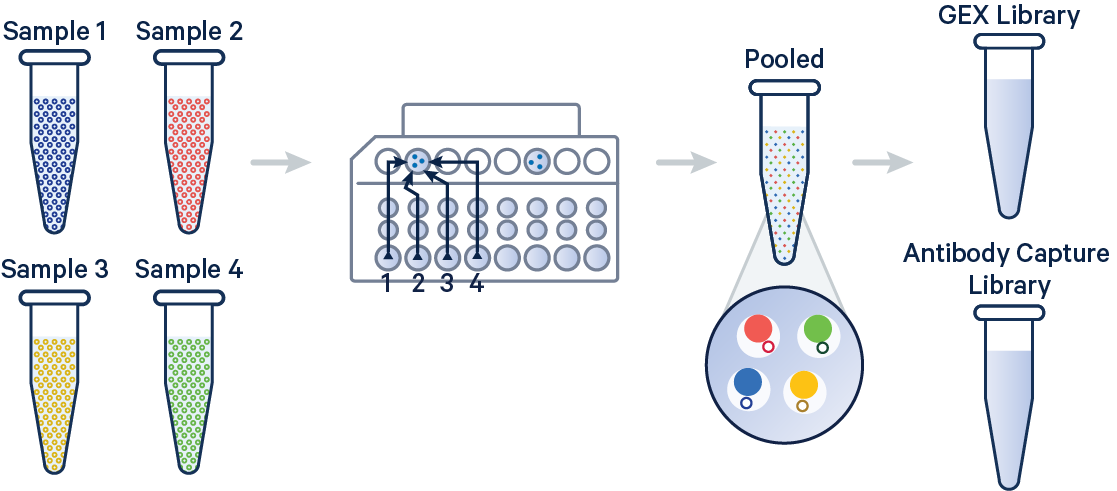
The config below specifies how to handle this setup using the cellranger multi pipeline.
[gene-expression]
reference,/path/to/transcriptome
create-bam,false
[feature]
reference,/path/to/feature_ref.csv
[libraries]
fastq_id,fastqs,lanes,feature_types,
gex1,/path/to/gex_fastqs,Gene Expression
ab1,/path/to/ab_fastqs,Antibody Capture
[samples]
sample_id,ocm_barcode_ids
sample1,OB1
sample2,OB2
sample3,OB3
sample4,OB4
In this configuration, cellranger multi will generate separate output folders for each technical replicate (i.e., one output folder for each OB1, OB2, OB3, and OB4).
If you want to treat the technical replicates as a single sample and generate a unified output folder instead, you can modify the [samples] section like this:
[samples]
sample_id,ocm_barcode_ids
sample1,OB1|OB2|OB3|OB4
By combining all OCM barcodes under a single sample_id, the analysis will be processed as one sample, and only a single output folder will be created.
The output file structure for OCM libraries is similar to that of 3' CellPlex and is described on the Sample Multiplexing outputs page.
In this example, 5' Gene Expression, V(D)J-B (BCR), and Antibody Capture libraries were created from four samples that were multiplexed on-chip. Technical replicates are identified by different OCM barcodes (for 5', OB1 = blue, OB2 = red, OB3 = green, and OB4 = yellow).
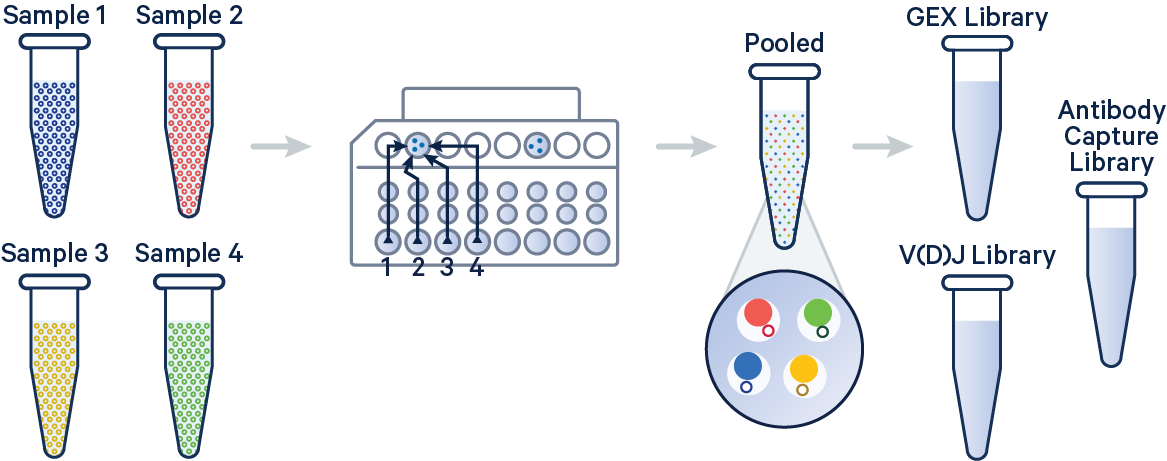
The config below specifies how to handle this using the cellranger multi pipeline.
[gene-expression]
reference,/path/to/transcriptome
create-bam,false
[vdj]
reference,/path/to/vdj_reference
[feature]
reference,/path/to/feature_ref.csv
[libraries]
fastq_id,fastqs,lanes,feature_types,
gex1,/path/to/gex_fastqs,Gene Expression
VDJ1,/path/to/vdj_B_fastqs,VDJ-B
ab1,/path/to/ab_fastqs,Antibody Capture
[samples]
sample_id,ocm_barcode_ids
sample1,OB1
sample2,OB2
sample3,OB3
sample4,OB4
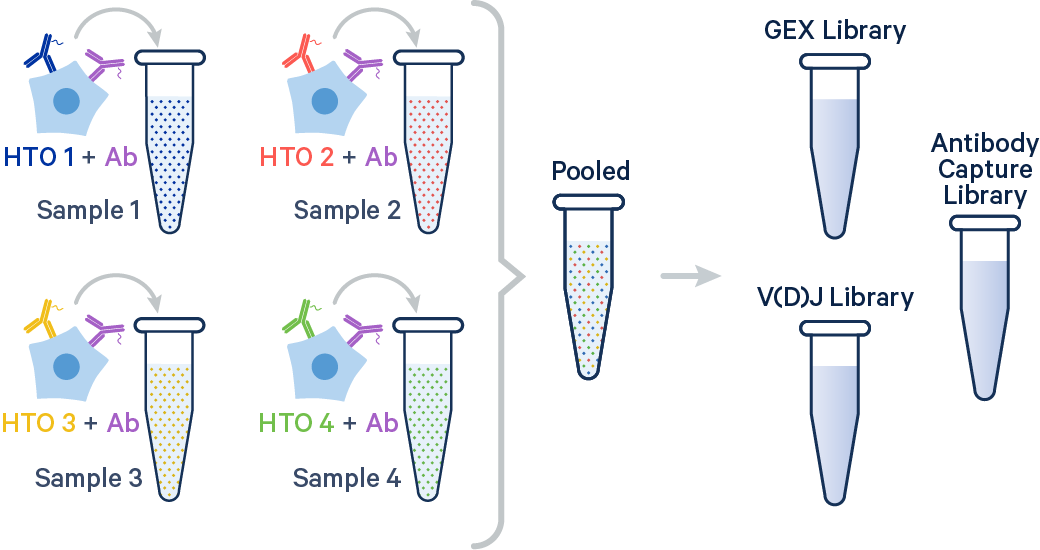
Cell or sample multiplexing with Antibody Capture (non-OCM hashtag oligos) is enabled starting from Cell Ranger v9.0. However, please be aware that this pipeline is not officially supported by the 10x Genomics Technical Support team. Additionally, hashing on OCM and hashing with Flex are both disabled in Cell Ranger.
To demultiplex samples hashed with Antibody Capture, both a Gene Expression library and an Antibody Capture library are required. Without Gene Expression data, Cell Ranger cannot demultiplex hashed samples. If gene expression profiles are not of particular interest, you may perform shallow sequencing. A suggested starting point for sequencing the GEX library is 5,000 reads per cell. However, the minimum Gene Expression sequencing depth needed for accurate cell calling may vary based on the sample type and data quality. Please read the 10x Genomics Technical Note CG000148 to learn about how sequencing depth and cell number influence the detection of major cell types in peripheral blood mononuclear cells (PBMCs).
Some additional advantages to including a Gene Expression library:
- Including a Gene Expression library, even with shallow sequencing, can help troubleshoot when library or sample quality is poor.
- T or B cell barcodes with incomplete rearrangements, which may occur in certain leukemias or developmental stages, are often excluded from the call set when the cell calling algorithm relies solely on the V(D)J library. However, these barcodes are retained in the Gene Expression library.
- Overall, the presence of a Gene Expression library enhances the characterization of cells and improves the robustness of data evaluation.
The oligonucleotide sequences used for sample demultiplexing are specified in the Feature Reference CSV and linked to individual samples in the samples section of the multi config CSV.
An example Feature Reference CSV for hashing with Antibody Capture is available on the Feature Reference CSV page. For a complete list of input files required to process libraries hashed with Antibody Capture, visit the Inputs for multi section on the List of inputs page.
This section provides example multi config CSVs for setting up cell or sample hashing using Antibody Capture. The samples section requires the hashtag_ids field for demultiplexing. For more details, visit the multi config CSV documentation.
In this example, two samples were multiplexed using TotalSeq-B™ antibodies from BioLegend.
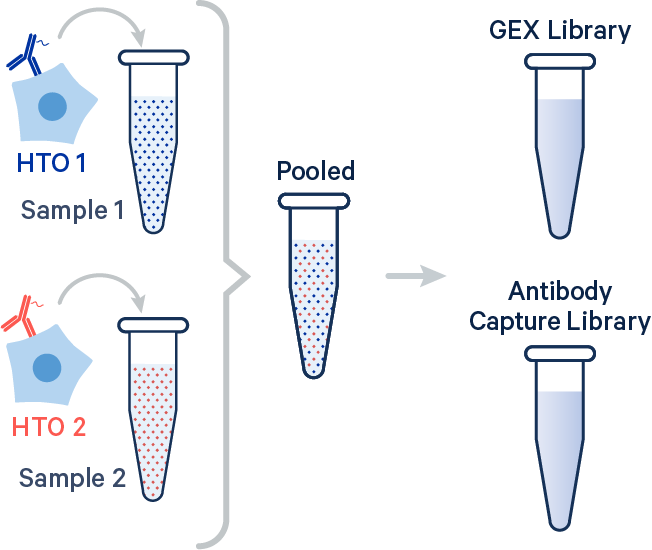
[gene-expression]
reference,/path/to/transcriptome
create-bam,true
[feature]
reference,/path/to/feature_ref.csv
[libraries]
fastq_id,fastqs,feature_types
gex1,/path/to/gex_fastqs,Gene Expression
ab1,/path/to/ab_fastqs,Antibody Capture
[samples]
sample_id,hashtag_ids
Sample1,TotalSeqB_Hashtag_1
Sample2,TotalSeqB_Hashtag_2
The output file structure for samples hashed with Antibody Capture is similar to that of 3' CellPlex and is described on the Sample Multiplexing outputs page.
In this example, four samples were multiplexed using TotalSeq-C™ antibodies from BioLegend.
[gene-expression]
reference,/path/to/transcriptome
create-bam,true
[vdj]
reference,/path/to/vdj_reference
[feature]
reference,/path/to/feature_ref.csv
[libraries]
fastq_id,fastqs,feature_types
gex1,/path/to/gex_fastqs,Gene Expression
vdj,/path/to/vdj_fastqs,VDJ
ab1,/path/to/ab_fastqs,Antibody Capture
[samples]
sample_id,hashtag_ids
Sample1,TotalSeqC_Hashtag_1
Sample2,TotalSeqC_Hashtag_2
Sample3,TotalSeqC_Hashtag_3
Sample4,TotalSeqC_Hashtag_4
Cell Ranger 6.0 and later supports analyzing 3' Cell Multiplexing data with the cellranger multi pipeline.
Here are a few example multi config CSVs for some common product configurations, along with simplified diagrams for the corresponding experimental set up. Replace /path/to with the absolute path to your data, and customize the text according to the experiment's sample, library, and file names. Ensure that the multi config is saved in CSV format with the CSV extension.
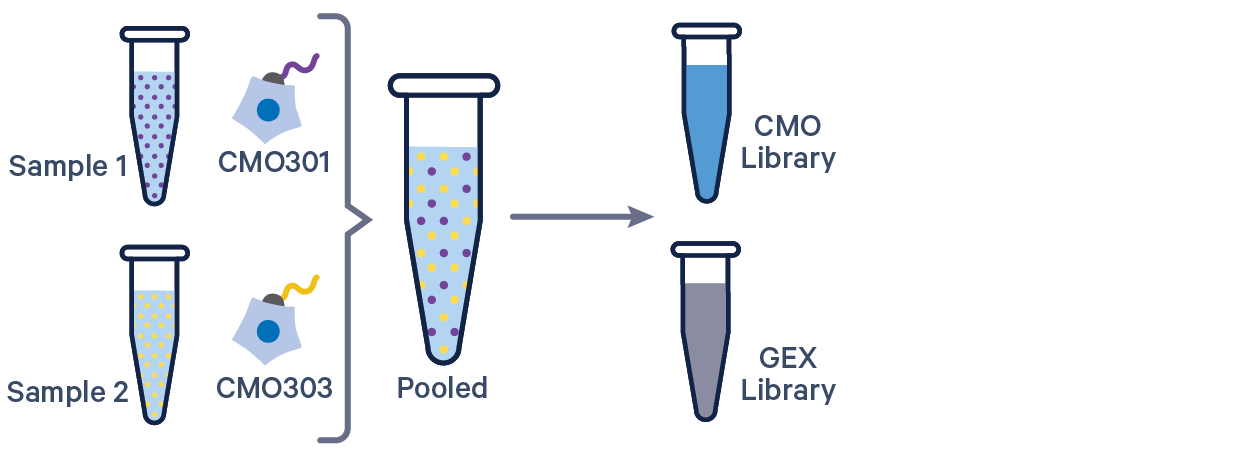
[gene-expression]
reference,/path/to/transcriptome
create-bam,true
[libraries]
fastq_id,fastqs,feature_types
gex1,/path/to/fastqs,Gene Expression
mux1,/path/to/fastqs,Multiplexing Capture
[samples]
sample_id,cmo_ids
sample1,CMO301
sample2,CMO303
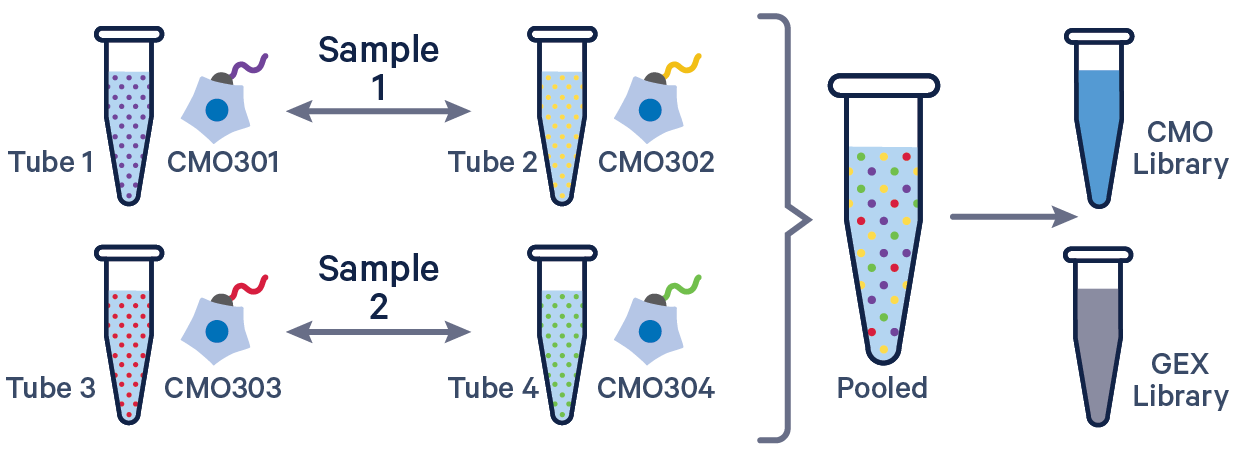
See example dataset. Note usage of the | to separate CMO tags. Learn more about when to use multiple CMOs per sample here.
[gene-expression]
reference,/path/to/transcriptome
create-bam,true
[libraries]
fastq_id,fastqs,feature_types
gex1,/path/to/fastqs,Gene Expression
mux1,/path/to/fastqs,Multiplexing Capture
[samples]
sample_id,cmo_ids
sample1,CMO301|CMO302
sample2,CMO303|CMO304
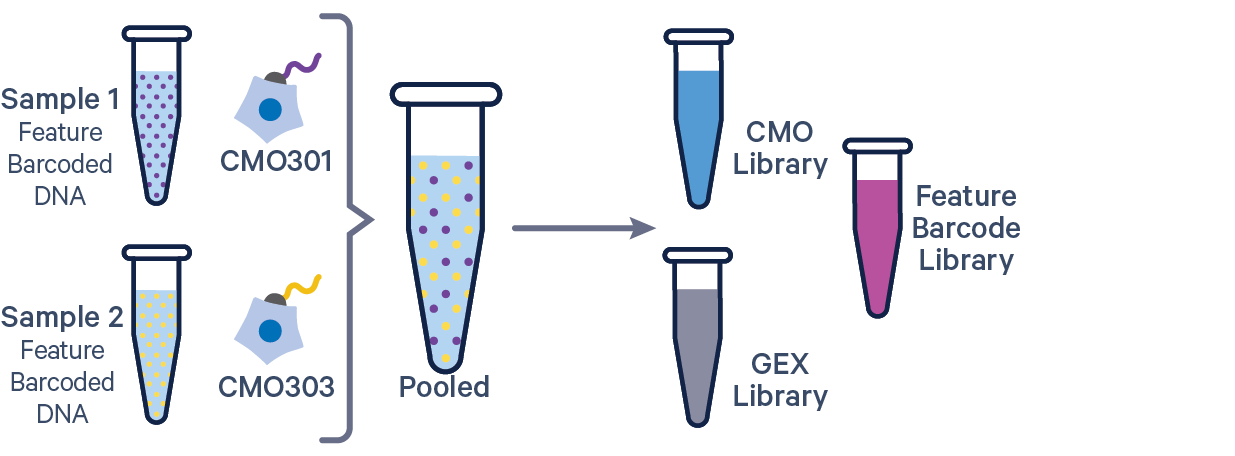
The additional Feature Barcode library in this config CSV example ([libraries] section) is Antibody Capture. Specify CRISPR Guide Capture as the feature_types for CRISPR Feature Barcode experiments.
[gene-expression]
reference,/path/to/transcriptome
create-bam,true
[feature]
reference,/path/to/feature_reference.csv
[libraries]
fastq_id,fastqs,feature_types
gex1,/path/to/fastqs,Gene Expression
abc1,/path/to/fastqs,Antibody Capture
mux1,/path/to/fastqs,Multiplexing Capture
[samples]
sample_id,cmo_ids
sample1,CMO301
sample2,CMO303
The cmo-set option in the [gene-expression] section of the multi config CSV allows you to provide a reference for custom Cell Multiplexing oligos. The design of this reference is nearly identical to the Feature Barcode Reference used to describe Feature Barcodes, with one difference: the feature_type is required to be Multiplexing Capture instead of those feature types supported in the Feature Barcode reference.
The id column may contain alphanumeric, underscore, and hyphen characters. Special characters are generally prohibited, except for the pipe (|) character which can only be used to separate multiple CMO IDs from the same sample in config CSV.
For example, Cell Ranger's default CMO reference looks like this (built into Cell Ranger):
id,name,read,pattern,sequence,feature_type
CMO301,CMO301,R2,5P(BC),ATGAGGAATTCCTGC,Multiplexing Capture
CMO302,CMO302,R2,5P(BC),CATGCCAATAGAGCG,Multiplexing Capture
CMO303,CMO303,R2,5P(BC),CCGTCGTCCAAGCAT,Multiplexing Capture
CMO304,CMO304,R2,5P(BC),AACGTTAATCACTCA,Multiplexing Capture
CMO305,CMO305,R2,5P(BC),CGCGATATGGTCGGA,Multiplexing Capture
CMO306,CMO306,R2,5P(BC),AAGATGAGGTCTGTG,Multiplexing Capture
CMO307,CMO307,R2,5P(BC),AAGCTCGTTGGAAGA,Multiplexing Capture
CMO308,CMO308,R2,5P(BC),CGGATTCCACATCAT,Multiplexing Capture
CMO309,CMO309,R2,5P(BC),GTTGATCTATAACAG,Multiplexing Capture
CMO310,CMO310,R2,5P(BC),GCAGGAGGTATCAAT,Multiplexing Capture
CMO311,CMO311,R2,5P(BC),GAATCGTGATTCTTC,Multiplexing Capture
CMO312,CMO312,R2,5P(BC),ACATGGTCAACGCTG,Multiplexing Capture
The default CMO reference above is available as a downloadable CSV.
The barcode-sample-assignment option in the [gene-expression] section of the multi config CSV allows users to provide a file that manually specifies the barcodes for each sample. It will override Cell Ranger's default cell calling and tag calling steps, and may be useful in cases where data with microfluidic failures can be partially rescued. This feature allows users to import custom tag calling done via 3rd party tools as well (see the Tag assignment of 10x Genomics CellPlex data using Seurat's HTODemux function Analysis Guide for help).
Here is an example multi config CSV:
[gene-expression]
reference,/path/to/transcriptome
barcode-sample-assignment,/path/to/barcode_sample_assignment.csv
create-bam,true
[libraries]
fastq_id,fastqs,feature_types
gex1,/path/to/fastqs,Gene Expression
mux1,/path/to/fastqs,Multiplexing Capture
[samples]
sample_id,cmo_ids
sample1,CMO301
sample2,CMO303
The barcode-sample CSV file has at most two columns, one for the barcode sequence and another that is either the sample ID or the tag assignment. A barcode can only be assigned to one sample; barcodes with multiple sample or tag entries will result in an error in Cell Ranger. Here are two examples:
Option 1: Assign to samples
Barcode,Sample_ID
ACGTACGTACGTACGT-1,Jurkat
CGTACGTACGTACGTA-1,Raji
GTACGTACGTACGTAC-1,Jurkat
TACGTACGTACGTACG-1,Raji
...
Option 2: Assign to tags
Barcode,Assignment
ACGTACGTACGTACGT-1,CMO1
CGTACGTACGTACGTA-1,Multiplet
GTACGTACGTACGTAC-1,Blank
TACGTACGTACGTACG-1,Unassigned
...