Breaking ground in rare disease research with single cell sequencing

In honor of Rare Disease Day, February 29, 2020, we share several recent studies that utilized single cell sequencing to characterize the underlying pathological features of rare cancers and cystic fibrosis.
Scientists, doctors, patients, and families come together every year on the last day of February to raise awareness about diseases that don’t always get the most attention yet, nonetheless, have a significant impact in the lives of affected individuals. This is Rare Disease Day, February 29, 2020. According to the World Health Organization, there are approximately 7,000 recorded rare diseases and around 400 million people worldwide suffer from them (1).
These statistics raise several questions: What is a rare disease? Where are we in identifying the underlying mechanisms for these rare diseases? How close are we to treatments? The WHO classifies rare diseases as those that affect less than one person out of 2,000 (1). These diseases impact all areas and systems of the human body, and include some very rare cancers. Examples of rare diseases include cystic fibrosis, a genetic disease that causes the mucus in a person’s lungs or digestive system to dangerously accumulate, and Hutchinson-Gilford Progeria syndrome, a genetic condition that leads to accelerated aging in children. Indeed, an estimated 80% of rare diseases are of genetic origin (1), meaning they are driven by some genetic loss, gain, or mutation.
This factor, coupled with the frustrating gap in knowledge for most rare diseases, points to a need for basic research that yields a deeper understanding of the underlying biology of the diseases, including connections between pathological features at the molecular, cellular, and whole-system levels. The following studies offer some promising insights into the origins and features of a few of these rare diseases, and also demonstrate the discovery power of technology that reveals the genomic and transcriptomic character of biological systems impacted by disease at single cell resolution.
New lung cell type advances our understanding of cystic fibrosis
A new epithelial cell type, discovered through single cell analysis, may be responsible for the disease symptoms of cystic fibrosis. Cystic fibrosis is a disease that affects the lungs and other mucus-lined passageways in the body. An abnormally thick, sticky mucus accumulates in these passageways as a consequence of mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which controls production of a protein responsible for moving chloride and sodium ions across cell membranes (2). CFTR is expressed in cells that line the airways and other affected glands and tracts in the body. However, these tissues are heterogeneous, making it difficult to determine the underlying cellular culprit(s).
Daniel Montoro and colleagues from Massachusetts General Hospital and the Broad Institute resolved this uncertainty when they performed single cell gene expression profiling on murine trachea tissue. They identified seven cell types, including one novel population that represented only 1% of airway epithelial cells and had characteristics similar to salt-balancing ionocytes in fish gills or frog skin. Differential gene expression analysis revealed that these cells, labeled pulmonary ionocytes, were the primary source of CFTR gene activity (3). This discovery could have major implications for treatment of cystic fibrosis, and has already begun to redirect research on gene therapy for CFTR mutations.
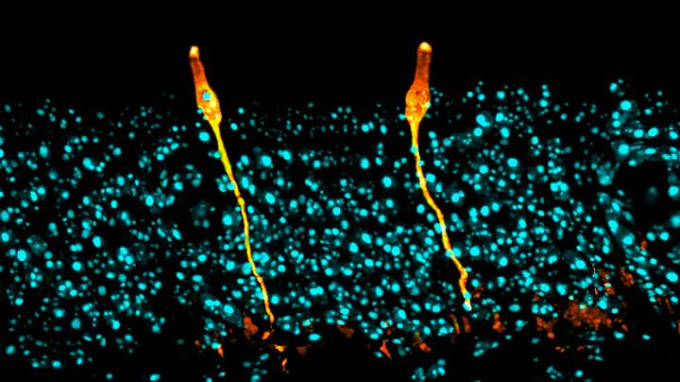
You can explore this study in an article out from the Broad Institute. 10x Genomics guest author Jennifer McArthur also wrote about this study in an article titled, Discovering Rare Cell Types with Single Cell RNA-Seq.
Macrophages may influence therapeutic resistance of rare child brain tumors
The interaction between macrophages and cells of a rare tumor appear to drive tumor heterogeneity and recurrence. Atypical teratoid/rhabdoid tumors (ATRT) are an aggressive class of cancers that affect the central nervous system in young children. Most ATRT tumors show somatic, that is, un-inherited, gene mutations in SMARCB1, a gene suppressor that encodes a protein involved in chromatin remodeling (4). This encoded protein is also known as INI1: it is known to act as a “tumor suppressor,” protecting cells from turning into cancer, and is often silenced in rare cancers like renal medullary carcinomas and ATRT (5).
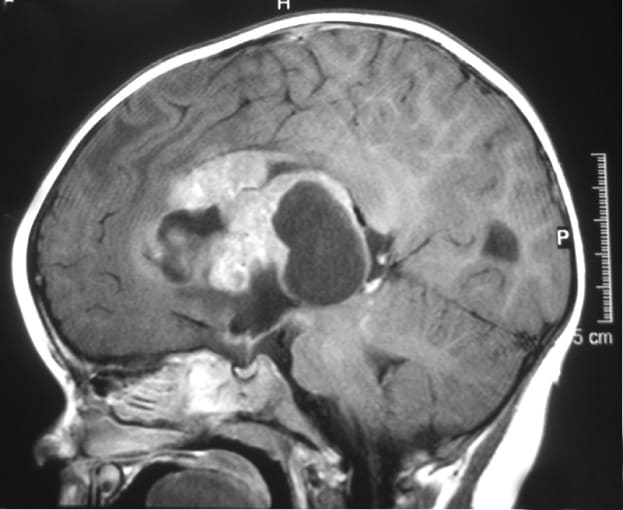
Historically, scientists have been stumped by the poor prognosis for patients with ATRT and unpredictable response to treatment. In an effort to understand what biological features drive disease severity, and find predictive biomarkers, Victoria Melcher and colleagues from the University Children’s Hospital in Münster, Germany, used immunofluorescence staining to study the immune cell infiltrate of 34 primary human ATRT tissue samples. Through this analysis, they identified a prominent population of heterogeneous CD68+ cells. To further distinguish the role and prevalence of CD68+ cells between subgroups of ATRT (ATRT-SHH and ATRT-MYC), they used single cell RNA-sequencing to study fresh tumor tissues derived from genetically engineered murine models. scRNA-seq enabled identification of marker genes for each subgroup and also revealed the infiltration of CD68+ macrophages specifically in the ATRT-MYC subgroup.
This finding led the team to question how CD68+ macrophages were interacting with tumor cells in the ATRT-MYC subgroup, and what influence this interaction had on prognosis. Melcher and her team developed an orthotopic xenograft mouse model using cells derived from a SMARCB1-negative human tumor and performed scRNA-seq again on the xenograft. They identified SMARCB1-negative tumor clusters and macrophage clusters, then performed read alignments with human and murine reference transcriptomes on these cells. While the macrophage cluster was SMARCB1-positive, and so clearly of murine origin, some of the cells of the tumor cluster contained murine genetic information. Specifically, some of these tumor cells expressed murine macrophage transcripts, including CD68, indicating that the macrophages from the mouse host had exchanged genetic information with human tumor cells. Gene expression analysis of this resulting intermediary cellular phenotype revealed that these cells overexpressed genes known to mediate chemo-resistance and associated with tumor recurrence and metastasis (6).
This study ultimately demonstrates the utility of single cell RNA-sequencing to unmask previously hidden features in rare diseases, including subgroup heterogeneity, critical gene expression profiles, and cell-type-specific interactions that drive disease severity and therapeutic resistance.
Uncovering a possible immune checkpoint blockade target for a rare eye cancer
A comprehensive single cell analysis of the tumor microenvironment of uveal melanoma has revealed a possible therapeutic target for the once poorly understood disease. A subtype of ocular melanoma, uveal melanoma arises in the uveal tract of the eye, the layer of pigmented tissue found beneath the white of the eye. This tissue is composed of melanocytes (pigment cells), muscle, and blood vessels, and cancer typically arises by transformation of melanocytes (7). The exact cause of this transformation into cancer cells is unclear, but scientists have found a few genes that occur more frequently in individuals with ocular melanoma. About 50% of patients have a loss of genetic material in chromosome 3, while others have a mutated form of the gene BAP1, a tumor suppressor gene that is known to slow cell division, repair damage to DNA, and initiate apoptosis (7).
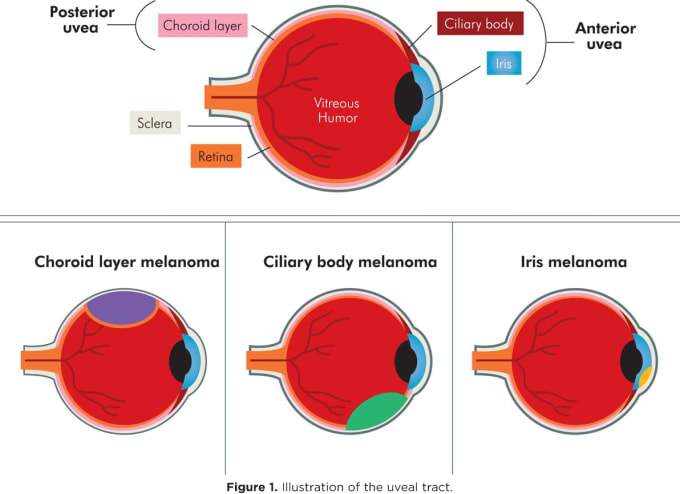
In an effort to better understand the underlying causes of this disease and why it tends to be unresponsive to immunotherapy, Michael Durante and colleagues from the Bascom Palmer Eye Institute at the University of Miami Miller School of Medicine performed single cell RNA-sequencing on eight primary and three metastatic human tumor samples, comprising 59,915 tumor and non-neoplastic cells. This revealed a clear division between two classes of tumor: class 1, BAP1 wild-type, and class 2, BAP1 mutant, the latter showing greater tendencies for metastasis. An investigation into the respective gene expression of tumor cell clusters and immune infiltrate clusters also showed that, of the 12 genes identified in a previous bulk study of uveal melanoma, five were expressed predominantly in tumor cells, six were expressed in both tumor and immune cells, and one, SATB1, was expressed predominantly in T cells. In this way, single cell analysis enabled an accurate stratification of the gene expression dynamics within the complex and heterogeneous tumor microenvironment.
Further analysis of the tumor samples using single cell copy number variation (CNV) technology allowed the team to characterize some of the ongoing genomic changes that drive tumor evolution. They found that one subclone of tumor cells, showing loss of heterozygosity in chromosome 3, still corresponded to class 1 primary tumors, indicating that mutation of BAP1 on the other copy of chromosome 3 may be necessary to drive the tumor to class 2 and further metastatic severity. With this perspective on the subclonal diversity of the tumor samples, and their scRNA-seq data, the team used Monocle, a software that enables transcriptional cell-state trajectory analysis, to identify 16 tumor cell states that clearly stratified between class 1 and class 2 tumor types. However, these states were distributed across subclones and cell cycle phases, suggesting the cells have transcriptional plasticity that may contribute to metastasis and therapy resistance.
The team then performed V(D)J recombination analysis of the immune cells from their tumor samples. Single cell RNA-sequencing analysis had revealed that CD8+ cytotoxic and T effector memory cells were present in all samples, however this deeper analysis into the immune repertoire showed that only three tumors, all class 2, had clonally expanded T cell populations. These T cells were exhausted, meaning they showed signs of losing their effector functions, and were expressing exhaustion-associated immune checkpoint molecules like LAG3, but had very low expression of PD-1 and CTLA-4. This finding suggests that low expression of traditional checkpoint molecules may contribute to the ineffectiveness of checkpoint blockade immunotherapies for uveal melanoma, and points to LAG3 as a potential novel target to alleviate T cell exhaustion (8).
The consensus of the rare disease community is clear: more research is necessary to understand rare diseases, to help inform patients and families, and to ultimately develop effective treatments. With single cell sequencing technology, researchers are uncovering critical disease insights more quickly and with more robust genomic, transcriptomic, and cellular information than ever before.
For more resources about these and other rare diseases, including links to patient organizations and clinical trials, please refer to the very thorough orpha.net, NORD, and OMIM databases.
References
- S Weely, and HGM Leufkens. Update on 2004 Background Paper, BP 6.19 Rare Diseases. 12 Mar. 2013, www.who.int/medicines/areas/priority_medicines/BP6_19Rare.pdf.
- https://rarediseases.org/rare-diseases/cystic-fibrosis
- D Montoro, A Haber, M Biton et al., A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature. 560, 7718 (2018).
- https://www.orpha.net/atypical-teratoid-rhabdoid-tumor
- https://rarediseases.org/rare-diseases/renal-medullary-carcinoma
- V Melcher, M Graf, M Interlandi et al., Macrophage-tumor cell interaction promotes ATRT progression and chemoresistance. Acta Neuropathologica. Published Online. DOI: 10.1007/s00401-019-02116-7. (2020).
- https://rarediseases.org/rare-diseases/ocular-melanoma
- M Durante, D Rodriguez, S Kurtenbach et al., Single-cell analysis reveals new evolutionary complexity in uveal melanoma. Nat Comm. 11, 496. (2020).