Bridging the genome and transcriptome in study of lupus immune regulation

In honor of National Lupus Awareness Month, we share two innovative studies that lend new insights on the relationships between genomic variation and cell type–specific gene expression features of circulating immune cells in systemic lupus erythematosus (SLE). These findings will help determine the genetic basis of lupus and characterize the downstream cellular and molecular mechanisms that drive the autoimmune condition.
How do you diagnose and treat a disease that manifests differently in every patient? Systemic lupus erythematosus (SLE) is a highly heterogeneous autoimmune condition, causing widespread inflammation and tissue damage in affected organs, including joints, skin, brain, lungs, kidneys, and blood vessels (1). For that reason, it remains incredibly difficult for doctors and scientists to pin down a cause, necessitating deeper molecular characterization of the immune landscape in lupus—at greater scale.
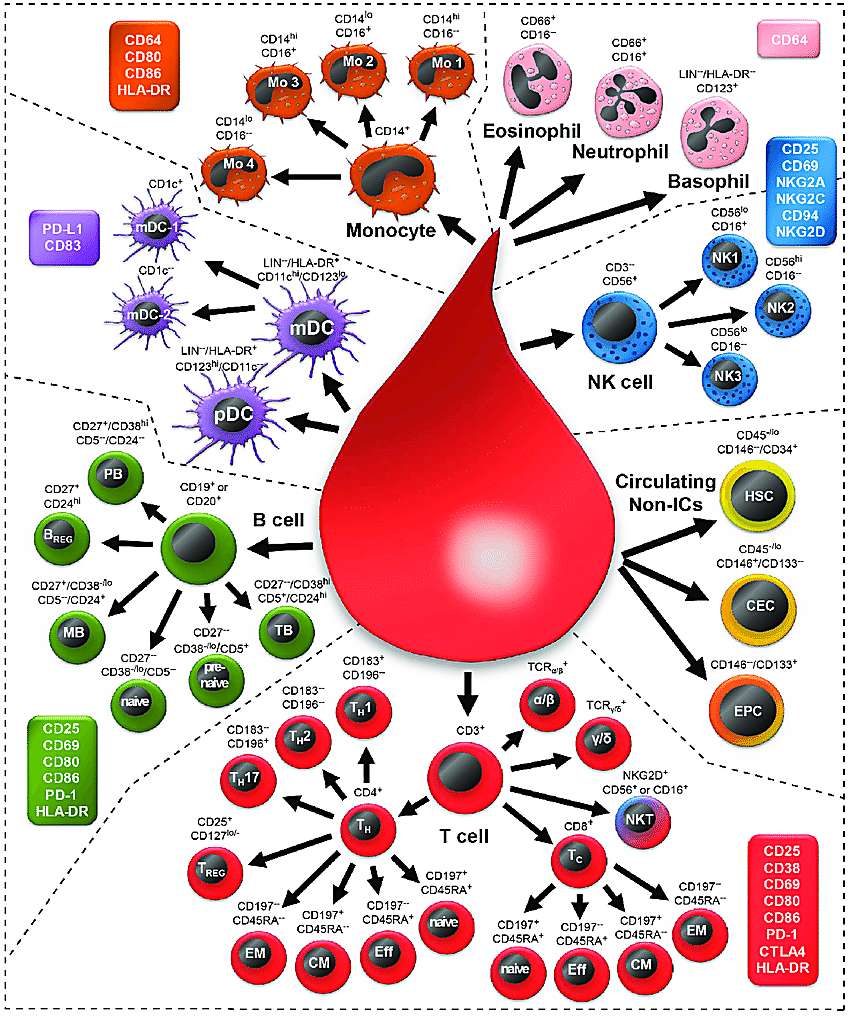
Two recent Science publications from Perez et al. and Yazar et al. described an experimental approach to study lupus immune regulation that promises great advances in our knowledge of the underlying mechanisms driving immune dysfunction, despite the diversity of clinical manifestations. Both approaches leveraged large patient cohorts and single cell RNA-sequencing technology to build molecular profiles of more than 1.2 million circulating immune cells from donor peripheral blood mononuclear cells (PBMCs). Specifically, Perez et al. profiled 162 lupus patient samples and 99 healthy controls, while Yazar et al. profiled a cohort of 982 healthy human subjects. Samples were genotyped to identify single nucleotide polymorphisms (SNPs) that could later be integrated with population genetics data to evaluate associations with autoimmune disease risk loci.
A comprehensive census of immune cells in lupus
Aberrant T-cell biology has been previously identified as a crucial culprit of lupus pathogenesis, “amplifying inflammation by secretion of pro-inflammatory cytokines, helping B cells to generate autoantibodies, and maintaining the disease through the accumulation of autoreactive memory T cells” (2). Yet a comprehensive atlas of the immune landscape in lupus has been lacking to date (3). This led Perez et al. to focus their efforts on characterizing the cellular composition and cell type–specific transcriptomic signatures of SLE and healthy samples, including, in SLE cases, disease-associated signatures that could represent a common phenotypic thread across all patients.
Perez et al. found that SLE PBMCs were marked by an overall decrease in percentage of CD4+ T cells, specifically a subpopulation of CD4Naïve cells, and an increase in classical monocytes. This may explain a phenomenon called lymphopenia—low white blood cell count—often observed in patients with SLE, which, importantly, was not the result of immunosuppressant treatment (3).
Surveying the CD8+ T-cell compartment in SLE cases, they observed, compared to controls, a significant increase in the percentage of granzyme-expressing CD8GZMH cells. These cells were transcriptionally heterogeneous, with mixed populations bearing elevated expression of cytotoxic, exhaustion, and interferon-stimulated gene (ISG) signatures. Single cell immune repertoire sequencing further refined the clonality of CD8+ T cells; of expanded clones, 59% were from CD8GZMH cells and 21% from CD8GZMK. Within the CD8GZMH subpopulation, cells expressing the cytotoxic gene expression signature were expanded at a ~4:1 ratio compared to cells expressing the ISG signature (3). Commenting on these T-cell features, the authors remarked, “the function of granzyme-H is not well characterized, but previous work demonstrated its divergent roles in initiating caspase-dependent apoptosis in T cells while initiating caspase-independent apoptosis in NK cells… The significant clonal expansion of GZMH+ CD8+ T cells, specifically within the cytotoxic subpopulation, suggests a pathogenic role for these cells in SLE.”
Focusing on the global differences between the lupus and healthy control immune landscapes, Perez et al. performed differential gene expression analysis for each of the 11 identified circulating immune cell types. This revealed 302 genes which were differentially expressed in at least one cell type between SLE cases and controls. Of these, 238 were previously undescribed differentially expressed genes in adult SLE and 56 were myeloid-specific (3).
This analysis also solidified six unique gene expression modules—such as Panup, a module that upregulated a group of ISGs across all cell types, and Myeup, a myeloid-specific module expressing IFITM3, which was previously classified in pediatric SLE. Importantly, modules could be used to stratify SLE cases from controls and, within cases, stratify patients enriched for disease flares. Taken together, these findings mark a huge leap in our foundational knowledge about the molecular characteristics that define the lupus immune landscape and varied clinical manifestations.
When population genetics meets single cell RNA-seq
Having defined the cell type–specific expression features of the peripheral immune landscape in lupus patients and healthy subjects, the research teams looked to integrate their findings with a deeper layer of biology: the genome. Could they find the underlying disease-associated genetic variants that resulted in these observed cellular and transcriptomic features of autoimmune disease?
Their approaches leveraged genotyping data that yielded a readout of expression quantitative trait loci (eQTLs), genomic loci that explain variation in mRNA expression levels. A local or cis-eQTL is one located in a noncoding region near the gene of origin, the gene that produces the transcript. According to Sumida and Hafler, “there are many common allelic variants in noncoding regions that have small effect sizes with complex interactions that are highly cell type and cell state dependent” (4), making it all the more essential to have a cell type–specific gene expression readout to map the variant back to.
Within the eight most prominent cell types from their SLE samples, Perez et al. identified 535 genes with at least one cell type–specific cis-eQTL and 1,207 genes with shared cis-eQTLs (2). Then they integrated SLE GWAS data with these loci to identify cell types where cis-eQTLs may harbor disease-relevant GWAS associations. This revealed that classical monocytes and B cells were the most enriched for GWAS-identified SLE variants, further reinforcing the role these cells may play in disease pathogenesis. More broadly, this revealed seven total SLE disease-associated loci colocalizing with cell type–specific cis-eQTLs, including 17q21, a locus previously linked to asthma, Crohn’s disease, and type 1 diabetes (3).
Yazar et al. took a population scale approach, integrating single cell eQTL data from their large sample cohort with GWAS data for seven common autoimmune diseases, including lupus. This revealed that 19% of cis-eQTLs shared the same causal locus as a GWAS risk association, and pointed to 305 specific loci that purportedly contribute to autoimmune disease through changes in gene expression in specific cell types and subsets. This included a number of shared causal loci, 38.4% of which are outside the major histocompatibility complex region (5).
These analyses highlight the potential of combining genomic and transcriptomic techniques to inform each other and more deeply annotate disease-associated risk loci, mapping genetic variation to not only causal genes, but the specific cell types and pathways that drive pathogenesis. With this high-resolution view of the underlying mechanisms involved in autoimmune diseases such as lupus, there is hope for improved diagnosis and treatments over time.
References:
- https://www.cdc.gov/lupus/facts/detailed.html#sle
- Suárez-Fueyo A, et al. T cells in systemic lupus erythematosus. Curr Opin Immunol 43: 32–38 (2016). doi: 10.1016/j.coi.2016.09.001
- Perez R, et al. Single-cell RNA-seq reveals cell type-specific molecular and genetic associations to lupus. Science 376: eabf1970 (2022). doi: 10.1126/science.abf1970
- Sumida T and Hafler D. Population genetics meets single-cell sequencing. Science 376: 134–135 (2022). doi: 10.1126/science.abq0426
- Yazar S, et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 376: eabf3041 (2022). doi: 10.1126/science.abf3041