Identifying ultra-broad coronavirus neutralizing antibodies from human blood samples with single cell multiomics

Editor's Note: Barcode Enabled Antigen Mapping (BEAM) is no longer available, but you can still analyze antigen specificity using compatible tools. Learn more here.
Alpha. Beta. Gamma. Delta. Omicron. The COVID-19 pandemic taught us all too well that viruses are constantly changing, and that variants—with their increased virulence, altered clinical manifestations, and ability to escape natural or vaccine-induced immunity—represent a serious threat to public health (1,2).
But researchers are taking steps to prevent the recurrence of our experiences with the COVID-19 pandemic by searching for broadly neutralizing antibodies against SARs-CoV-2 variants that can be used to make variant-resistant vaccines.
New tools to mine the human immune repertoire for antigen-specific B cells, bearing therapeutically desirable antibodies, are dramatically accelerating this search. Bryan Briney, PhD, Associate Professor of Immunology and Microbiology at Scripps Research Institute, has put these tools to the test. He recently led a groundbreaking research collaboration with 10x Genomics scientists to recover and characterize two broadly neutralizing antibodies against a diverse range of coronaviruses (3) using a single cell multiplexed antigen screening approach now commercialized as Barcode Enabled Antigen Mapping (BEAM).
But that’s just two out of the over 9,000 antigen-specific B cells and paired, full-length B-cell receptors Dr. Briney and his team identified in blood samples taken from convalescent COVID-19 patients. We explore their experiments and findings, as well as the game-changing implications for antibody discovery, vaccine development, and infectious disease research, in this article.
*Jump ahead to learn how BEAM helps recover full-length, paired antibody sequences at transformative scale, how the team applied multiplexed antigen screening to find broadly neutralizing antibodies against SARs-CoV-2, and what’s next for Dr. Briney’s research into antibody language modeling, plus how he envisions BEAM will contribute to it.**
Overcoming the antibody discovery bottleneck
When it comes to antibody discovery research, the problem isn’t availability. It’s access. The human body contains a massive number of B cells and, through the power of V(D)J gene recombination, a similarly vast repertoire of unique B-cell receptors (BCRs).
“You have tens of billions of B cells in your body at any given time, all of them make antibodies, and every one of them, to a first approximation, makes a different antibody. Where is that information stored? It’s a million times bigger than your genome if you tried to add up [the sequences of] all of those different antibodies,” said Dr. Briney, in his lecture at 10x Genomics headquarters.
Despite this potential, finding and characterizing the right antibodies, against a specific antigen of interest, and at a resolution that yields actionable insights, isn’t so easy. One reason is due to the paired nature of BCRs, each of which contains a heavy and light chain.
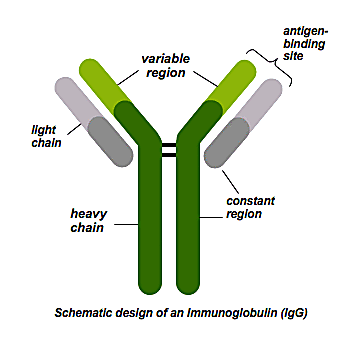
“The heavy and light chains of an antibody are different—they’re on different chromosomes, different genes, they’re totally different. If you want to get a full antibody molecule sequence, you have to do it at the single cell level. You can’t just take a bunch of B cells and sequence all the heavy chains, and all the light chains, and figure out which ones went together—there’s no way to know. The only way of doing it is through a single cell approach,” said Briney.
To achieve this resolution, Briney’s team used to perform a plate-based method where they would sort individual B cells into a 96-well plate, one cell per well, and do single cell PCR. “It was super expensive, super labor intensive, and we would be ecstatic if we got a dozen—or maybe even, heaven forbid, a hundred—antibodies that were interesting,” he said.
But single cell sequencing approaches have fundamentally changed the throughput and efficiency at which his team can now find these antibodies.
Using the Barcode Enabled Antigen Mapping workflow for antigen-specific B-cell discovery (BEAM-Ab), Briney and his team can now systematically interrogate hundreds of millions of B cells to sort and isolate cells with specificity against barcoded antigen assemblies. These assemblies could contain a variety of antigens of interest to enable multiplexed antigen screening in the same sample. Antigen-specific B cells are then analyzed via the single cell immune profiling workflow, providing a readout of each B cell’s full-length, paired receptor sequence and whole transcriptome.
“Now, instead of getting 100 antibodies we care about, we’re getting tens of thousands, potentially, of antibodies that we care about. It has exploded what we’re able to do,” Briney said.
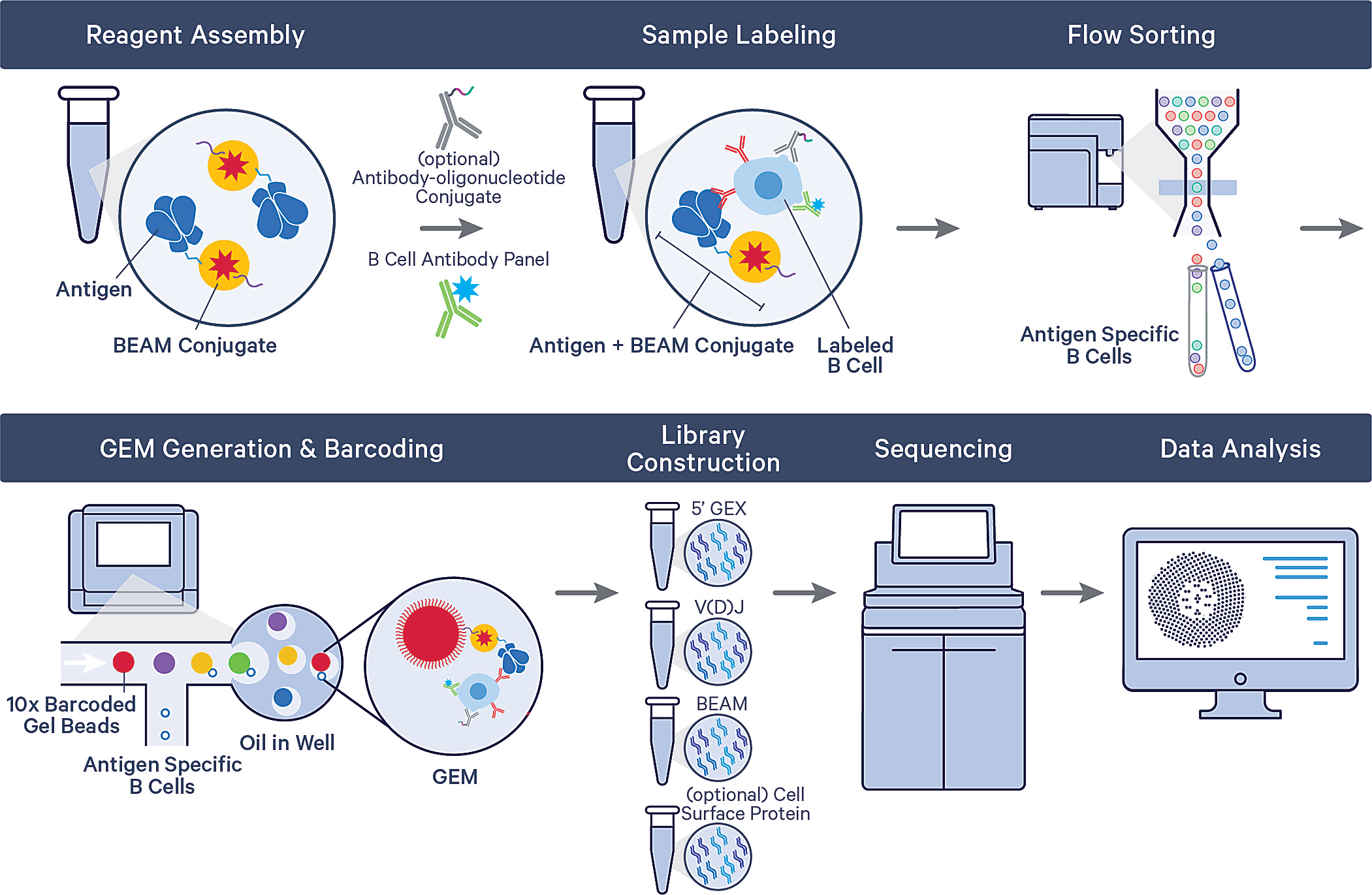
One experiment, one sample—2,737 mAbs against SARs-CoV-2
In their recent bioRxiv preprint (3), Briney’s team from the Scripps Research Institute along with a group of 10x Genomics scientists described an experiment using barcoded antigens and high-throughput single cell sequencing to identify potent antibodies against SARS-CoV-2. They took a sample of 108 peripheral blood mononuclear cells (PBMCs) from a recovered COVID-19 patient, then labeled B cells with a multiplexed antigen panel consisting of three uniquely barcoded antigen assemblies bearing either the prefusion trimeric Washington strain SARs-CoV-2 spike protein (WA1-S), prefusion trimeric D614G spike protein, or a human serum albumin (HSA) negative control.
After sorting antigen-specific B cells and running these cells through the full assay on the Chromium platform, they recovered 2,737 paired mononuclear antibodies (mAbs), then selected a subset of 239 to generate recombinant antibodies. Of this chosen subset, 106 (44%) neutralized the WA1 strain of SARS-CoV-2. Further testing against SARS-CoV-2 variant and seasonal coronavirus S-proteins revealed that one antibody, coined TXG-0078, was not only potently neutralizing against SARS-CoV-2, but also showed broad efficacy, binding to seasonal coronaviruses and all variant spike proteins except Omicron.
Interestingly, TXG-0078 bound to the N-terminal domain (NTD) of spike proteins; typically, the most potent antibodies against SARs-CoV-2 bind to the receptor-binding domain (RBD) of the spike protein. While there are other NTD-targeting antibodies that neutralize SARs-CoV-2, TXG-0078 is the only known mAb against the NTD supersite with binding breadth that spans this diverse range of coronaviruses (3).
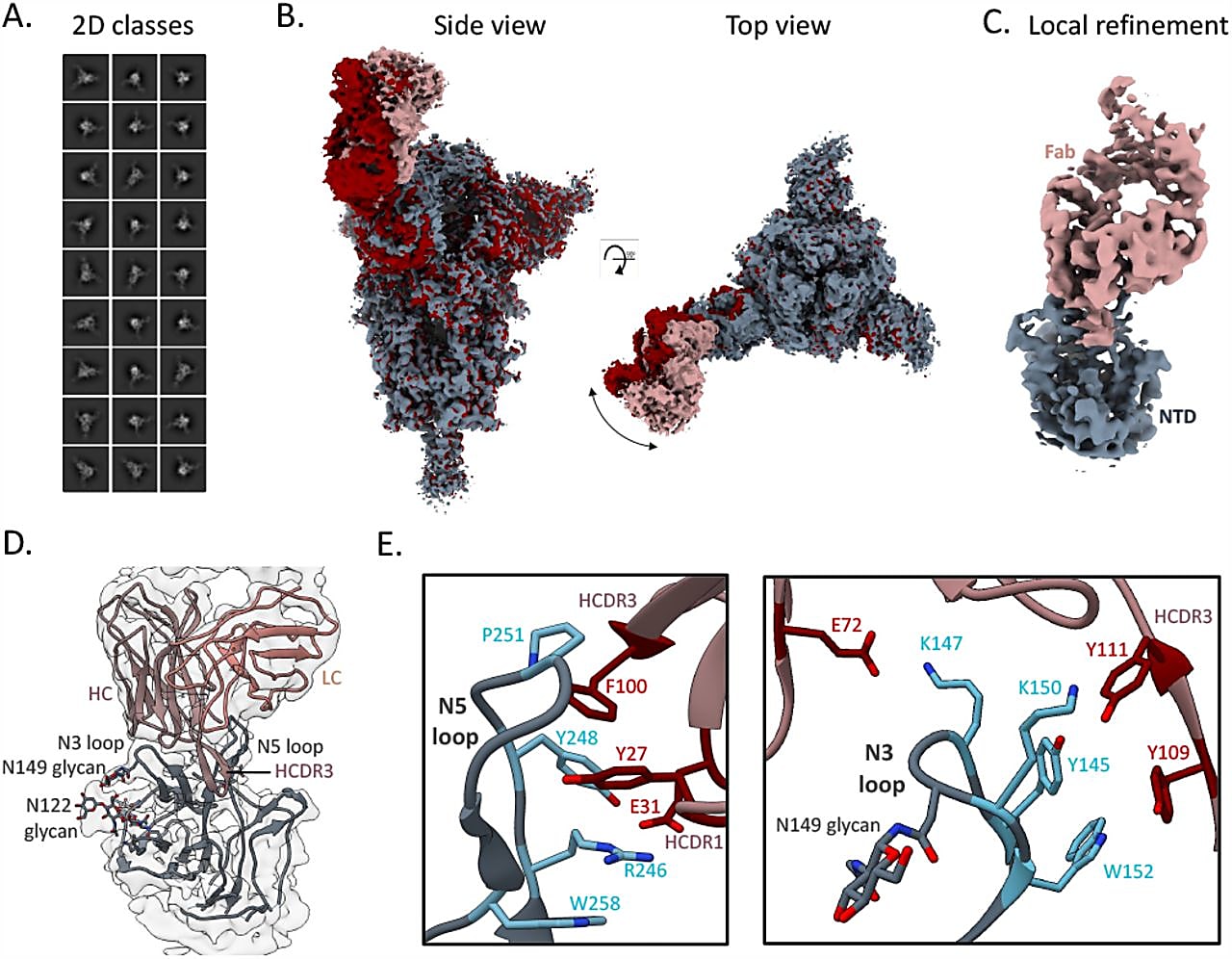
A good problem to have
It’s important to note that the TXG-0078 antibody was one select protein out of hundreds and even thousands of antibodies for which the team did not generate a recombinant protein or further validate. With this previously unimaginable scale and throughput by which to access potentially therapeutically relevant antibodies, immunologists have a good problem to solve now—namely, how to prioritize the right antibody sequences to make into recombinant proteins for downstream validation.
Data analysis tools built for the BEAM workflow, such as Loupe V(D)J Browser, can be used to visualize antigen-specific antibody sequences based on antigen specificity scores—a readout comparing UMI counts associated with antigens of interest against UMI counts associated with the control antigen. This helps identify the most valuable antigen-specific clonotypes. Some additional criteria may be needed to narrow down this selection even further, including evidence that antibodies are highly mutated (an indicator of maturity and possibly better binding potential through rounds of mutation) or perhaps have a shared theme of sequence, according to Briney.
Commenting on these selection criteria, Briney said, “In cases where we use multiple barcoded antigens in BEAM-Ab antigen assemblies, we can look for things that are the most interesting. If we’re looking for the most broad antibody, we can prioritize our selection by the antibodies that bind to the most antigens we put in there. We can also look for other things. For example, if two or three or four people make very similar antibodies, that suggests this antibody may be conserved across a lot of people. Those are potentially interesting. But we have no way of doing it that we know is foolproof. I would say there’s probably a 90% chance that there’s a better antibody in there that we haven’t found.”
A second broadly neutralizing antibody, CC24.2, makes a mean cocktail
Briney’s team dug deeper into the available human immune repertoire and found even more broadly neutralizing SARs-CoV-2 antibodies by screening blood samples from four convalescent donors who were infected early in the pandemic, then vaccinated later. They simultaneously broadened their antigen panel to include S-proteins from the WA1, beta, and gamma strains of SARS-CoV-2, as well as SARS-CoV S-protein and the RBD from a bat coronavirus closely related to SARS-CoV-2 (3). This experiment recovered 6,302 paired mAbs. One antibody, in particular, CC24.2, was broadly neutralizing against SARS-CoV and a diverse range of SARS-CoV-2 variants, including Omicron.
Given the complementary specificities between CC24.2 and TXG-0078, the team tested the prophylactic benefits of these two antibodies in an in vivo mouse model. They treated mice with 300 μg of mAb in four treatment groups, including TXG-0078 alone; CC24.2 alone; a two-mAb cocktail consisting of 150 μg each of TXG-0078 and CC24.2; and ZIKV-1, a negative control targeting Zika virus. Mice were then challenged with the WA1 strain of SARS-CoV-2, revealing that TXG-0078 alone and the antibody cocktail provided the best protection (3).
Explaining the implications of these results, Briney said, “If the antibody is there, SARs-CoV-2 does not make you sick. That’s the gist of it.”
These results ultimately demonstrate that there’s more valuable antibody content, hidden within human samples, that the scientific community has yet to discover. The broader therapeutic implications are clear, as Briney noted, “60 to 70% of drugs in development now are antibodies, not small molecules. A lot of them are mouse antibodies that have been humanized. There are a lot of development issues, because you’re putting something that’s not human into something that is human. If we isolate antibodies from humans, those will immediately be better candidates for drugs. We know they’re safe in humans, and they’re targeting viruses in a way that actual survivors of these diseases target viruses. Using this approach to discover antibodies from humans can accelerate the drug development process.”
Antibody breadth increases with SARs-CoV-2 vaccination
Briney’s team made some additional discoveries about the evolving immune response to repeated SARs-CoV-2 exposure through their single cell analysis of antibody serum in convalescent COVID-19 patients.
Briney said, “One thing we noticed was that after vaccination, the serum of these individuals was broader, with better ability to bind a diverse array of coronaviruses. What happened? What did the vaccine do that made these antibodies broader, or that recruited new antibodies to add to the breadth of the serum?”
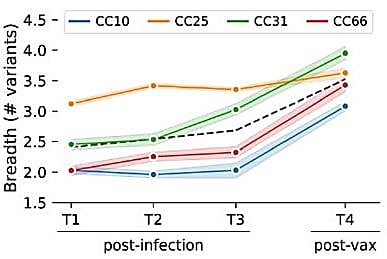
With the single cell resolution insights provided through the BEAM workflow, his team could answer that question. They observed that this increase in antibody binding breadth was associated with a class switch towards the IgG isotype, and a greater number of mutations in the antigen-binding regions of individual antibodies, both of which suggested a more mature antibody response as a result of vaccination (3).
“Your breadth of variant protection is increasing because the antibodies themselves are increasing in cross-reactivity, not because you're recruiting new antibodies that just happen to be specific to more variant epitopes,” said Briney.
“This high-resolution serology approach allows us to deconvolute the patterns of breadth in your antibody repertoire at the individual antibody level. It’s the equivalent of going from bulk RNA-seq to single cell RNA-seq.”
Training the future of antibody discovery
Single cell interrogation of antigen-specific B cells and their full-length, paired receptor sequences, at massive scale not only promises to change the way we do antibody discovery research today, but in the future as well. Referencing the leaps in innovation we’ve seen through the implementation of machine learning algorithms to train language models, Briney drew a parallel to antibody discovery, suggesting that antibody language models could aid prediction of the best antibody sequences with which to generate recombinant proteins—and, potentially, much more.
Speaking on the potential of these antibody language models, Briney said, “They could generate antibody sequences de novo. So you could ask it to give you a flu antibody sequence, and it could make it for you. You could give the model a sequence and do sequence optimization.” Briney’s team is working to train these antibody language models, but they need specific input data to make the model do what they hope it will do someday. That is, full-length, paired antibody sequences.
“There is something, a pattern between those heavy and light chains, that we haven’t been able to determine yet,” Briney explained. “There is some sort of mutual information between the antibody chains that are being paired that we can leverage to make a much better model.”
That’s where the data provided by BEAM-Ab is changing the game, and where the ability to scale single cell analysis to millions, even hundreds of millions of B cells, could make a transformative contribution to the development of accurate antibody language models and, in the end, better therapeutic antibodies.
To learn more about Bryan Briney’s research into SARs-CoV-2 antibody discovery, please explore his preprint →
References:
- Paul P, et al. Genomic surveillance for SARS-CoV-2 variants circulating in the United States, December 2020–May 2021. MMWR Morb Mortal Wkly Rep 70: 846–850 (2021). doi: 10.15585/mmwr.mm7023a3external
- Siddiqui S, et al. Recent chronology of COVID-19 pandemic. Front Public Health 10: 778037 (2022). doi: 10.3389/fpubh.2022.778037
- Hurtado J, et al. Deep repertoire mining uncovers ultra-broad coronavirus neutralizing antibodies targeting multiple spike epitopes. bioRxiv (2023). doi: 10.1101/2023.03.28.534602