Sequence with confidence: understand index hopping and how to resolve it

Index hopping occurs when reads from an RNA-seq library are assigned to the wrong sample. Though it’s usually a rare phenomenon, index hopping can introduce deleterious artifacts into single cell RNA-seq experiments. Now, with dual indexing for all 10x Genomics transcriptomic libraries, you can approach your single cell and spatial experiments with greater confidence. Read on to explore dual indexing and how it mitigates these sequencing challenges, and learn more in our Technical Note.
We know index hopping can negatively impact sequencing data, but where does it come from? How does index hopping happen, and how do sequencers unintentionally enable it?

Inside a NovaSeq sequencer lies a patterned flow cell covered with billions of nanowells. Each well is covered with a lawn of P5 and P7 oligos that hybridize to specialized adaptors on the oligonucleotide fragments of, for example, your single cell RNA-seq library. Amplification of library fragments occurs simultaneously with template hybridization to the nanowells in a process called ExAmp, or exclusion amplification. Amplification happens much faster than hybridization; as soon as a single fragment hybridizes to a seeding oligo in a nanowell, it will take over, amplifying until all the available oligos are bound, thereby excluding any other templates. This results in a high proportion of monoclonal clusters across all nanowells and dramatically increased sequencing capacity. The NovaSeq enables more sequencing, faster, and at lower cost.
However, any sequencer using ExAmp can be sensitive to index hopping. Index hopping can occur whenever samples are multiplexed and may disrupt the correct assignment of cDNA sequence to sample index. Index hopping is caused by a partial library fragment annealing to a fragment in another nanowell, creating a chimeric molecule with a swapped sample index. Index hopping may result from unligated adaptors in solution, which can hybridize to the complementary sequence of the adaptor on the unbound end of a properly hybridized fragment and extend, adding the incorrect sample index to that sequence and creating a hybrid library in that nanowell. If the index-hopped strand then denatures and seeds another nanowell, it will generate a false signal, incorrectly assigning that read to a different sample during demultiplexing. Index hopping can also arise from long stretches of low diversity in cDNA sequence that allow one fragment to bind to another in a separate nanowell, as well as DNA breakage during amplification.
Though index hopping is rare for most applications, its impact can be quite large. In a paper published last month in Nature Communications by Rick Farouni et al., low levels of index hopping—less than 1%—could lead to phantom molecules that complicated downstream analyses of single cell RNA-seq experiments. Phantom molecules made up almost 10% of total UMIs for a quarter of the samples tested, and the impact was more severe for low complexity samples, where only a few unique molecules dominate the sequencing output. In addition to potential confounding effects on cell characterization and cell type identification, high levels of phantom molecules impacted cell calling and resulted in overestimation of cell number (1).
Fortunately, index hopping can be mitigated by making sure that unligated adaptors are cleaned up from your library with SPRIselect before sequencing—consult the Tips and Best Practices section of the Chromium Next GEM Single Cell 3’ v3.1 (Dual Index) User Guide for guidance in removing excess adaptors. Additionally, you can mitigate index hopping with dual indexing, a method to eliminate sample misassignment due to sequencing errors.
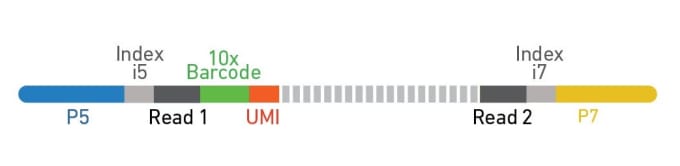
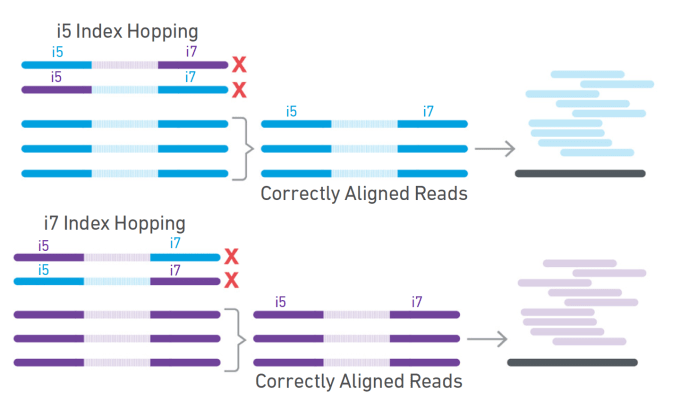
Dual indexing is now enabled with Single Cell Gene Expression v3.1 and Single Cell Immune Profiling v2, in addition to Visium Spatial Gene Expression. For dual index libraries, each library fragment contains two specific sample index sequences paired with the P5 (Figure 2, blue) and P7 (Figure 2, yellow) adaptors. Only specific combinations of sample indices are used, meaning that if index hopping does occur and a chimeric molecule is amplified, it can be computationally identified and discarded because the i5 and i7 sample indices won’t match (Figure 3). Typically, only 0.1–2% of reads are filtered out due to index hopping. If that sounds like searching for a needle in a haystack, don’t worry. Cell Ranger v4.0 will automatically search for and filter out index-hopped reads for you, so you don’t have to think about it.
Dual indexing also provides added convenience, since it has become the standard sequencing library configuration. For example, if you use a core facility for sequencing, your wait times can be reduced by multiplexing libraries that have the more common dual index format. With the advantage of accelerated batch sequencing, you can easily implement dual indexing with our Single Cell Gene Expression, Single Cell Immune Profiling, or Visium Spatial Gene Expression Solutions.
Get started with Single Cell Gene Expression v3.1 with Dual Indexing today: look for the dual index icon (Figure 4) in the margin of the User Guide, or consult this Technical Note for more information.
References
- R Farouni et al., Model-based analysis of sample index hopping reveals its widespread artifacts in multiplexed single-cell RNA-sequencing. Nat. Comm. 11, 2704 (2020).