index-hopping-filter is a tool that filters index-hopped reads from a set of demultiplexed samples. The tool detects and removes likely index hopped reads from demultiplexed FASTQs, and in turn emits new, filtered, FASTQs with similar file and directory layout as the inputs, suitable for use with cellranger count and cellranger vdj.
Note: Currently index-hopping-filter only supports samples produced by 3' Single Cell Gene Expression, 5' Single Cell Immune Profiling, Fixed RNA Profiling (FLEX), and Single Cell ATAC solutions. It cannot be used on Spatial Gene Expression samples.
Index hopping is a known phenotype that can impact any pooled samples sequenced on Illumina platforms utilizing both patterned flow cells and exclusion amplification chemistry (i.e. HiSeqX, HiSeq3000/4000, NovaSeq).
Below is some literature from Illumina outlining the background behind index hopping:
- Effects of Index Misassignment on Multiplexing and Downstream Analysis
- Minimize index hopping in multiplexed runs
To reduce and filter hopped sample indexes, we recommend the following library preparation best practices for any indexed sequencing library:
- Remove free adapters from library preps
- Ensure no sign of leftover primers or adaptors are present when performing post Library Construction QC (BioAnalyzer/TapeStation/Fragment Analyzer)
- Only pool libraries before sequencing
- A final 1.0X SPRI can be performed on these pooled libraries to remove any traces of free adaptors
- Use Unique Dual Indexes vs. Single indexed libraries or Combinatorial Dual Indexes - Unique Dual Indexes will be available for Single Cell Gene Expression, Single Cell Immune Profiling, and Spatial Gene Expression Products. Visium - Q4 2019, SC GEX and IP with full software support - mid 2020
10x created index-hopping-filter to reduce the number of index-hopped reads reaching downstream analysis. It detects, from whole flow cells, duplicate library molecules appearing in more than one sample. Reads determined to be so duplicated are removed. A sample where the molecule is present with a high read count indicates the molecule is from that sample, and reads of that molecule are not removed.
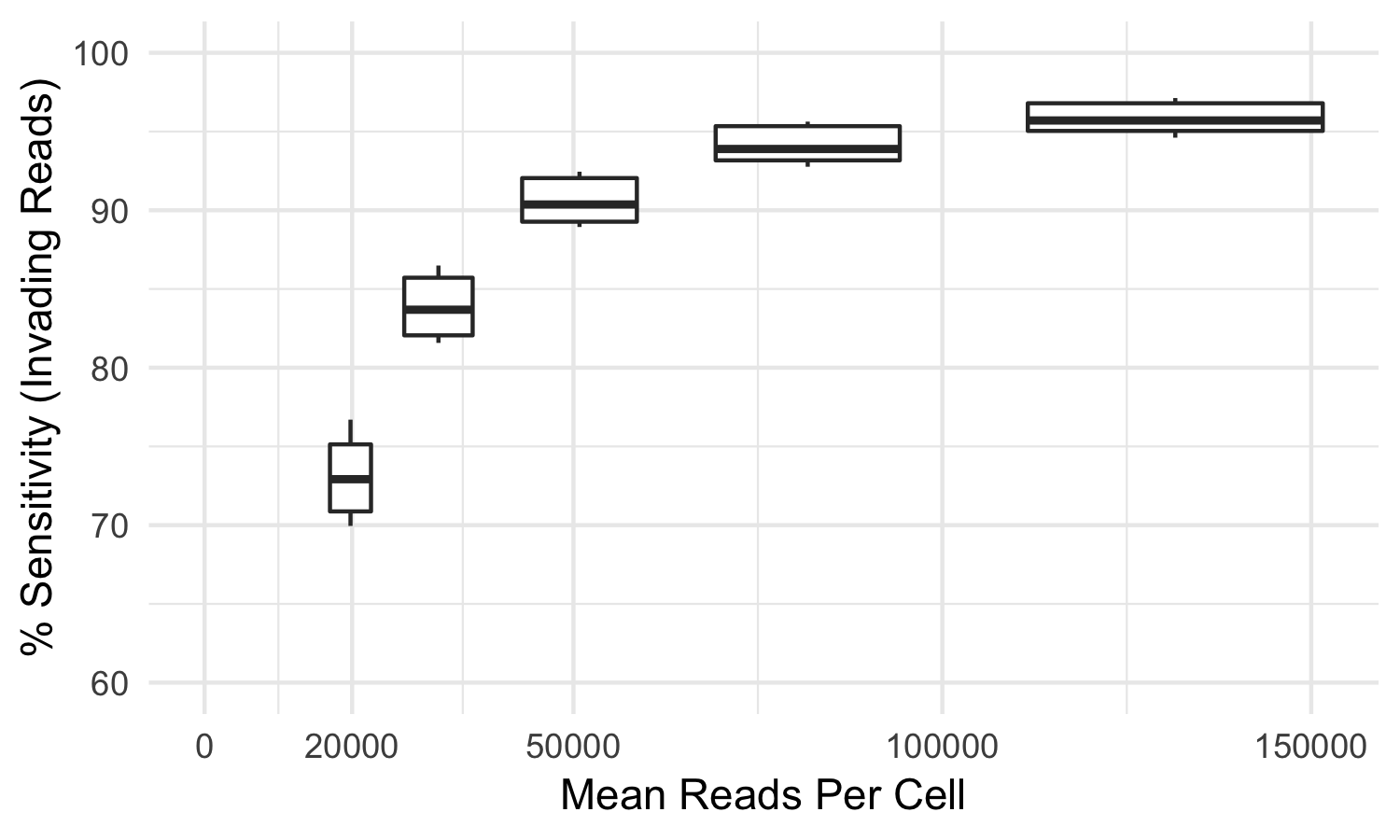
Some index hopped library molecules are only observed in incorrect samples and thus cannot be detected as index hopped. For this reason, the tool mitigates but cannot eliminate all index hopped reads. In our testing, using samples sequenced to the recommended sequencing reads per cell, index-hopping-filter removes >70% of index hopped reads compared to a dual-indexed ground truth. At lower sequencing depths, or if less than all samples from a flow cell are used, a smaller fraction of index hopped reads will be detected. The following graph depicts the sensitivity (over a whole flow cell) of index-hopping-filter to reads from invading barcodes compared to a dual-indexed dataset as a function of sequencing depth.
index-hopping-filter is available for Linux and is compatible with Redhat/CentOS 5.2 or later, and Ubuntu 8.04 or later.
Download index-hopping-filter v1.1
index-hopping-filter is a single executable that can be run directly and requires no compilation or installation. Place the executable in a directory on your PATH and make sure to chmod 700 to make it executable.
The mkfastq subcommand of cellranger produces FASTQ files directly usable by index-hopping-filter. If you have configured the IEM SampleSheet.csv or cellranger mkfastq simple CSV to uniquely identify distinct 10x libraries, then you can use the cellranger mkfastq output directory as input to index-hopping-filter filter.
Inputs of index-hopping-filter (10x Single Cell Gene Expression or Immune Profiling)
index-hopping-filter’s two subcommands both accept as input either a cellranger mkfastq output path or an index-hopping-filter configuration CSV. This CSV file must conform to the following schema, where SampleId must be an integer in the range [0, 65534].
| R1 | R2 | SampleId |
|---|---|---|
/path/to/sample1/R1.fastq.gz | /path/to/sample1/R2.fastq.gz | 1 |
/path/to/sample2/R1.fastq.gz | /path/to/sample2/R2.fastq.gz | 2 |
Inputs of index-hopping-filter (10x Single Cell ATAC)
index-hopping-filter’s two subcommands both accept as input either a cellranger mkfastq output path or an index-hopping-filter configuration CSV. This CSV file must conform to the following schema, where SampleId must be an integer in the range [0, 65534]. Compared to the configuration CSV for 10x Single Cell Gene Expression or Immune Profiling solutions, this configuration CSV requires an additional R3 column, and the --atac flag must be provided to the subcommand.
| R1 | R2 | R3 | SampleId |
|---|---|---|---|
/path/to/sample1/R1.fastq.gz | /path/to/sample1/R2.fastq.gz | /path/to/sample1/R3.fastq.gz | 1 |
/path/to/sample2/R1.fastq.gz | /path/to/sample2/R2.fastq.gz | /path/to/sample2/R3.fastq.gz | 2 |
Outputs of index-hopping-filter mkcsv
index-hopping-filter mkcsv produces a configuration CSV to stdout based on the input. Redirect stdout to a file (e.g. index-hopping-filter mkcsv /path/to/mkfastq/outs 1>config.csv) and modify it as needed.
Outputs of index-hopping-filter filter
| Outputs | Description |
|---|---|
| outs/fastq_path | Directory containing emitted FASTQs with suspect index hopped reads removed. |
| outs/web_summary.html | An HTML web summary that describes the number and percentage of reads removed per sample. |
| outs/filtered_reads.csv | A CSV file describing the starting number of reads and the number of reads removed for each SampleId. |
| _log | A log file for diagnostic purposes. |
index-hopping-filter mkcsv
| Option | Description |
|---|---|
-a, --atac | Generate a configuration csv for 10x scATAC-seq data |
-h, --help | Prints help information |
-v, --version | Prints version information |
index-hopping-filter filter
| Option | Description |
|---|---|
-a, --atac | Provide if you are running 10x scATAC-seq data |
-n, --nthreads | Maximum number of threads to use (default: number of cores) |
-h, --help | Prints help information |
-v, --version | Prints version information |
Product compatibility
index-hopping-filter currently only works with 10x Single Cell Gene Expression, 10x Single Cell Immune Profiling, and 10x Single Cell ATAC solutions. Future versions may include support for other products.
Multiple FASTQs from the same GEM wells
Correctly specifying SampleId is important. If multiple samples drawn from the same underlying 10x GEM well are passed into index-hopping-filter with distinct SampleIds, then index-hopping-filter will detect many reads as index hopped and remove them. This scenario arises when e.g. sequencing Immune Profiling-enriched libraries alongside the Gene Expression libraries from which they were derived. To avoid these false positive index hopping calls, use index-hopping-filter mkcsv to construct an initial configuration CSV, providing the cellranger mkfastq output directory as input. Next, modify the CSV to give demultiplexed samples derived from the same 10x GEM well a common SampleId value in the CSV.