A data tale: finding the story in your single cell gene expression data

We spoke with Eleanor Howe, PhD, Founder and CEO of Diamond Age Data Science, a Compatible Product Partner with 10x Genomics, to get her take on the best approaches to single cell gene expression experimental design and data analysis. Read on to learn how making connections as part of her research expanded her career and find expert tips and tools for single cell RNA-seq analysis to help you unlock the story of your data.
The marvelous challenge of scientific research is taking a complex biological sample—a tumor biopsy, PBMCs from a multi-patient cohort in a clinical trial, a cell line treated with different drug compounds—and accurately telling its story. What hidden insights does the sample contain? What can it teach us about cancer biology, or the efficacy of certain therapeutic candidates? As they seek to answer these questions, scientists become translators, interpreting biological facts and deriving crucial implications for how we understand and treat complex diseases through the language of data.
Diving deeper into biological complexity through data analysis can be challenging for a bench scientist or even an expert bioinformatician. But in the words of Eleanor Howe, PhD, Founder and CEO of Diamond Age Data Science, there is a tenacity required to unlock more insights from every sample. Regarding single cell gene expression data, she said, “...you're never going to just want a few things out of single cell, you're going to want everything. You're going to want to dig in, I promise.”

This article explores Dr. Howe’s journey of digging into data, finding the right statistical models to unlock deeper knowledge of biological complexity, and becoming a scientific partner to researchers in need of a bioinformatician. Read her data tale to learn how collaboration and people are as central to effective data analysis as the expert insights she shares about the differences between single cell RNA-seq, bulk RNA-seq, and flow cytometry readouts; how to properly model multiple-sample differential expression analysis; what tools to use for upstream and downstream analysis; and what the field needs going forward. Whether you’re just getting started, or are an experienced bioinformatician looking for advanced tips, there’s something here for you.
A bioinformatician at large: The path to Diamond Age
As a long-time computational biologist and staff scientist at the Center for the Development of Therapeutics at the Broad Institute, Dr. Howe developed a unique, insider perspective on the nuances of single cell gene expression experimental design and data analysis. She worked as an internal bioinformatics consultant—“I was kind of a bioinformatician at large”—supporting diverse projects and scientific teams, including several cancer and rare disease studies. One long-running project, a study of autosomal dominant inherited kidney disease, stood out in Dr. Howe’s experiences because of the great people, the cool statistics that harkened to her own PhD program, and the challenging data analysis job. “I didn't entirely know what the right model was that we needed to use in order to do this analysis…. I did the thing that you do when you are a consultant—and I started asking around, ‘Who is the right person to talk to about this kind of statistical problem?’”
Learning and teaching new analytical methods, and building connections between people, was at the heart of Dr. Howe’s work at the Broad. “I would go talk to a new set of people and figure out what they needed, and then I would either learn how to do the thing that they needed, or I would help them find the people to do the thing they needed. Or, I would go find the person who could teach me how to do the thing that they needed.”
This powerful combination of technical skills, people skills, and networking between her colleagues at the Broad and previous institution, the Dana-Farber Cancer Institute, ultimately prepared Dr. Howe for the next big step—founding Diamond Age Data Science, a bioinformatics consultation company. Recalling a time when she connected a new client to one of her former students at the Broad who was an expert in RNA splicing analysis, she said, “That was one of the things that convinced me that I could do this. I really liked the process of talking to somebody and figuring out what they needed, how to explain it to them, what they understood about bioinformatics and what they didn’t, figuring out what the project was, and then saying, ‘Yeah, I know somebody who can do that.’”
Dr. Howe is now putting her experience into practice as the leader of Diamond Age, which has recently become 10x Genomics’ first Data Analysis Partner. Dr. Howe and her team of expert bioinformaticians specialize in analyzing single cell RNA-sequencing data for their biotech and pharmaceutical clients, including common downstream analytical methods like modeling differential gene expression, identifying cell types, and performing trajectory analysis. Beyond single cell analysis and other, more standard whole genome and exome analysis, the team can also build custom analytical pipelines. “We build the kind of software that you need a bioinformatician to build. We don't build the kind of software that a more traditionally trained software engineer would build. If you need someone with a PhD to design the thing properly and then make sure it works every single time, that's the kind of pipeline and infrastructure builds that we do.”
Explore more of the services Diamond Age provides here.
An expert’s approach to single cell gene expression data analysis
Dr. Howe’s path to consulting demonstrates that no matter your level of experience in bioinformatics, partnering with other scientists can help you overcome roadblocks in data analysis and unlock the secrets of your samples. In that spirit of collaboration, Dr. Howe shared some of her experience with us. Find five expert insights about how to approach single cell gene expression experiments and data analysis in the sections below.
1. Single cell RNA-seq data is different from a flow cytometry readout.
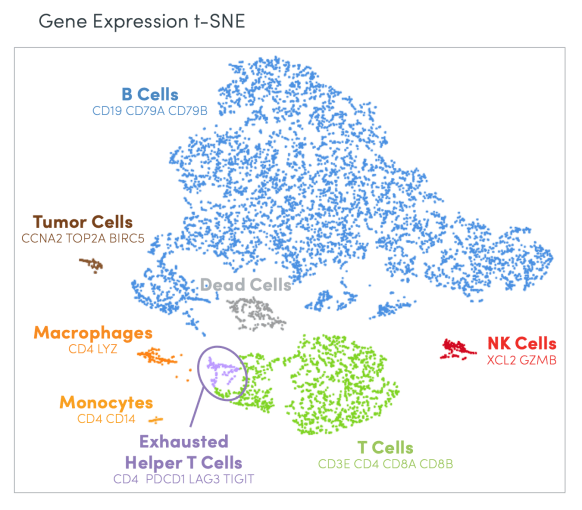
Dr. Howe directed her first insight to scientists who may be more accustomed to the bench, but want to get started with analyzing single cell sequencing data. For this group of scientists, flow cytometry is likely a familiar tool, providing a readout of the presence or absence of a small set of cell surface proteins using antibodies to annotate and pool specific cell types. Dr. Howe emphasized the differences between this protein readout and the whole transcriptome readout from single cell RNA-seq, recommending that researchers use the entire transcriptome to assess cell identity instead of the typical one or two markers from flow sorting. “Start with a few gene markers to try to get a feel for which cluster is the cell type you care about, then look at the whole transcriptome from that cluster, and use that whole transcriptome profile to identify cells.”
As a complementary tip, Dr. Howe suggested, “Do a lot of reading with this technology. It's different from anything else, and it's worth investing the time in understanding how it works because it is so different. It's so powerful that you might be tempted to kind of skim it and then say, ‘Oh, this is good enough, I just need to get a few things,’ but you're never going to just want a few things out of single cell, you're going to want everything.”
Get started on your reading with some of these introductory resources.
2. Single cell RNA-seq data is different from a bulk RNA-seq readout.
Dr. Howe directed her next recommendation to experienced computational biologists, making an important distinction between single cell technology and bulk methods. “Single cell is not just a simple evolutionary change from bulk RNA-seq. It's a totally different thing, and the data has orders of magnitude more complexity to it and more value to it.” One of the differences between single cell and bulk in Dr. Howe’s experience is the analysis pipeline. Following a bulk study, researchers will typically do quality control, differential expression analysis, “then, maybe you do some pathway analysis. It's pretty straightforward, and you can do that with pretty low manual intervention.” In contrast, Dr. Howe noted that single cell analysis is more comprehensive because it’s marked by a number of additional decision points. “The analysis process is more dependent on the tissue type you're looking at, the cell types that you're interested in, and the exact research question that you're after. Are you interested in developmental trajectories of cells? Are you interested in RNA velocity to predict future states? Are you interested in differential expression between subpopulations of cells? Those are all very different questions that you can ask, and that you would have a much harder time asking with bulk, but you have all of these options with single cell. So, again, it's worth investing the time.”
3. Avoid the common pitfalls of multiple-sample differential gene expression analysis. Make the adjustment to your statistical model.
One of the pitfalls of treating single cell RNA-seq data as a bulk readout comes in the context of multiple-sample differential gene expression analysis. Dr. Howe recalled a project she worked on in which an experienced bioinformatician kept getting some extreme p-values. “He was doing differential expression of single cell data for a compound-treated and an untreated group of animals. They had a cell type they were interested in and they wanted to know what genes were affected by a compound, which is a common use case for bulk RNA-seq. The statistics for that are pretty straightforward. You just pop it in a differential expression tool, and off it goes. But with single cell, because you have multiple cells, you have multiple measurements on each animal. A first pass use of the tools that are available would probably cause those tools to treat each of those cells as an independent measurement. And that results in a p-value that is 10 to the minus 700, or something just completely ludicrous.”
Dr. Howe provided further context to this problem, explaining that “tools you can use off the shelf will sometimes treat each cell as equal to every other cell, regardless of how many cells came from each animal, or how many animals you have. They kind of lump all the cells from the treatment group with all the cells from the untreated group, regardless of which animal they came from, and just compare them all to each other.” In that case, the n, or total number of samples used for the statistical modeling of the comparison of the treated versus untreated group, is the number of individual cells, perhaps several thousand, rather than the number of unique samples, perhaps just three animals.
This inaccurate n is what creates the “funny results” that stump even bioinformaticians. However, Dr. Howe offered an approach that she uses to ensure appropriate treatment of the experimental design. “I used a mixed-effects model with an R package called MAST. This provides a way of grouping the measurements, the different cells from one animal, into a distribution for that animal. Then we compare those three distributions from the three animals in the treated group to the three animals from the untreated against each other. So your n is 6, and you end up with much more reasonable p-values.”
Dr. Howe clarified that the available tools to do differential gene expression analysis are not inherently flawed, but scientists can fall into the trap of using them inappropriately. “If you don't adjust your use of them in this way, your results will be incorrect. Most people, when they start doing single cell RNA-seq, they start with one sample. They want to look at one cell type and compare it to this other cell type. When you look at differential expression between those groups of cells, it’s appropriate to treat each cell as an independent measurement because you know intuitively that whatever p-value you get out of that comparison is really only relevant to the one sample you're looking at.” That p-value would allow a scientist to say with confidence there’s a difference between those annotated cell types in the sample. However, adding multiple samples to the mix complicates the model. Dr. Howe emphasized making the statistical adjustment to avoid reaching a false, generalized conclusion about the differences in cell populations between different samples.
4. Data quality matters. Do the pre-work to optimize sample preparation in order to get the best data.
Bioinformatics can only go so far. Dr. Howe acknowledged this, emphasizing the crucial role of sample preparation and maximum cell viability to ultimately get good data from your single cell gene expression experiment. “The data quality you get, of course, matters. I wouldn't rush through the optimization processes on the lab side. There is a little bit that we can do on the computer side to clean up stuff after the fact. There's a little bit, but not that much. So take the time to really get good at the cell prep, especially for difficult tissues.”
5. Use your full toolkit, in both upstream and downstream analysis.
Dr. Howe recommended a number of tools to conduct single cell gene expression data analysis, some pertaining to different stages of the analytical process. For the upstream processing, Dr. Howe said, “Cell Ranger is great. Most often, our clients come to us with data out of Cell Ranger, and we pick up and run from there.” For downstream processing, Loupe Browser is a great way to get initial insights for non-informaticians. Dr. Howe also recommends a combination of other well-known tools: “We like Seurat, we like Scanpy. Both of them are really powerful. They're open source. MAST is something that works well as an add-on with Seurat…. I commonly would use Seurat for the first half of the analysis—with the clustering, modeling, and batch correction—and then MAST for the differential expression, as kind of a pair…. We also use Monocle, for trajectory mapping.”
Looking to the future of single cell data analysis
Dr. Howe also offered some ideas about the next advancements the field will need for not only more efficient, streamlined single cell data analysis but also to empower researchers to explore the story of their datasets even more deeply. “I'm excited about the possibility that there will be tools for better exploration and presentation of the data, especially for folks to be able to do that internally. [...] Researchers need something that lets them explore their own datasets, alongside some public data or combined with all the other datasets they generated, but in a way that's easy for anyone to use.” Dr. Howe pointed to a browser tool or cloud-based analysis tools to enable this kind of data exploration. Single cell multiomic data integration is another innovation Dr. Howe is looking forward to: “The whole [Diamond Age] group is very excited about the integration of single cell RNA and single cell DNA data together. They want to see that, especially in tumor biology. That's going to be really amazing to see that at the same time.”
Another idea reflects on the current challenges of manual cell-type annotation. Dr. Howe remarked that “the marker-based identification of cells is kind of derived from the flow cytometry way of identifying cells. In some cases, that's the best we've got, and we can make it work. But it's not routinized; it's not reproducible, necessarily. I'm really looking forward to the day when we have a public resource for identifying cell types of all kinds...something that will help us put our data through a single cell RNA-seq pipeline, and out the other side have a pretty good assessment of which cells are in there and how many there are.” To reach that vision of an automated annotation process, Dr. Howe noted, would require more libraries, catalogs of marker genes, transcriptional profiles to identify characteristic cell types, and, at a minimum, a system to add to and organize this information.
Indeed, this vision points to the underlying complexity of biology that each data tale is seeking to unearth. As Dr. Howe put it, “I imagine that catalog will forever grow as we get more biology, more cell types, more cell subtypes, more disease states, more engineered cell types—I mean, who knows what, right?” Perhaps, beyond that, “we'll have a profound change in what we view as all cell types and decide that every single cell is unique, which is probably where we're headed.”
Anticipating this future for single cell research, marked by masses of increasingly granular data, we will continue to look to powerful data analysis tools and trusted collaborators to help us make the journey toward resolving biology and advancing human health. Many thanks to Dr. Howe for her insights and for her team’s work to support scientists in their search for the biological story in their single cell gene expression data.
For more information about the services Diamond Age Data Science provides, read their story. And, find further resources for single cell gene expression data analysis on our support site.
This article contains a discussion of analysis conducted by Dr. Eleanor Howe and colleagues. View and opinions do not constitute endorsement or promotion of 10x Genomics, Inc. or any of its products.