spaceranger mkfastq pipeline is deprecated and will be removed in a future release. Please use Illumina’s BCL Convert to generate Space Ranger-compatible FASTQ files. For detailed guidance, refer to the Demultiplexing with Illumina Software page.The Space Ranger workflow starts by demultiplexing the Illumina sequencer's base call files (BCLs) for each flow cell directory into FASTQ files. 10x has developed spaceranger mkfastq, a pipeline that wraps Illumina's bcl2fastq and provides a number of convenient features in addition to the features of bcl2fastq:
- Translates 10x sample index names into the corresponding oligonucleotides in the i7/i5 dual-index. For example, well A1 can be specified in the sample sheet as SI-TT-A1, and
spaceranger mkfastqwill recognize the i7 and i5 indices asGTAACATGCGandAGTGTTACCT, respectively. - Supports a simplified CSV sample sheet format to handle 10x use cases.
- Supports most
bcl2fastqarguments, such as--use-bases-mask. For a list of accepted arguments, see the Command Line Argument Reference, or runspaceranger mkfastq --help.
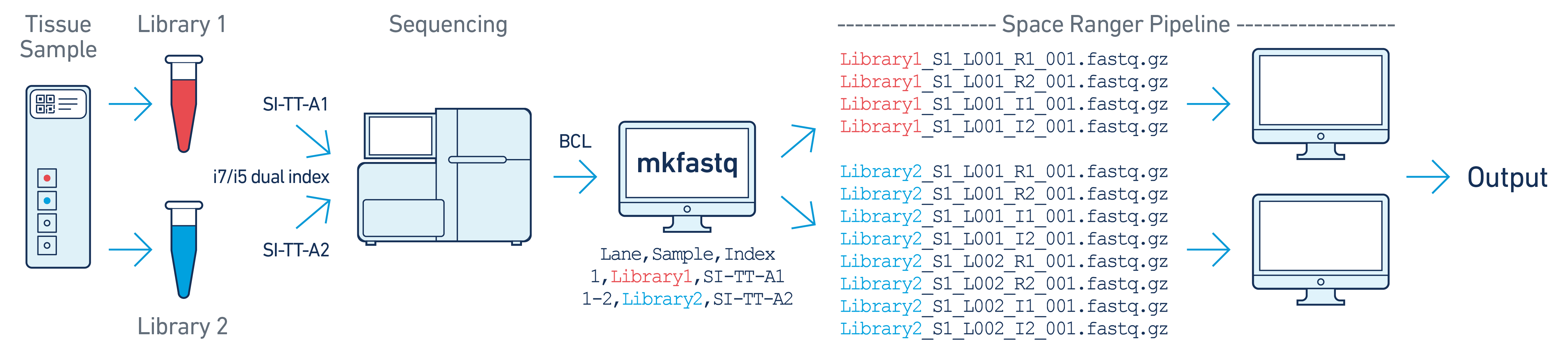
In this example, there are two 10x libraries (each processed through a separate capture area) that are multiplexed on a single flow cell. Note that after running spaceranger mkfastq, we run a separate instance of the spaceranger pipeline on each library.
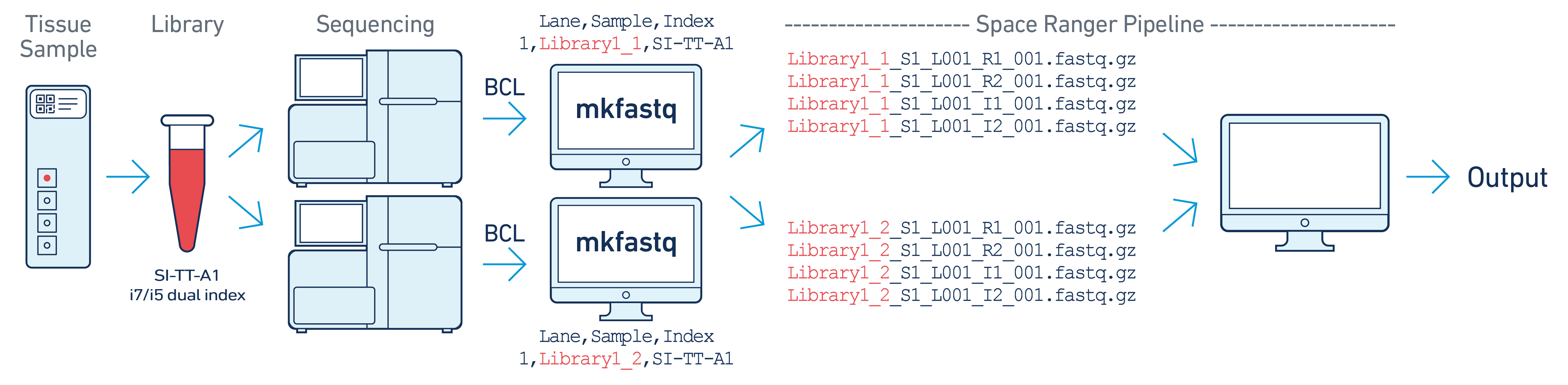
In this example, one 10x library is sequenced on two flow cells. Note that after
running spaceranger mkfastq, we run a single instance of the spaceranger
pipeline on all the FASTQ files generated.
spaceranger mkfastq recognizes two file formats for describing samples; a simple, three-column CSV format, or the Illumina Experiment Manager (IEM) sample sheet format used by bcl2fastq. The example below showcases running mkfastq with each format.
To follow along, do the following,
- Download the tiny-bcl tar file.
- Untar the tiny-bcl tar file in a convenient location. This will create a new
tiny-bclsubdirectory. - Download the simple CSV layout file: spaceranger-tiny-bcl-simple-1.0.0.csv.
- Download the Illumina Experiment Manager sample sheet: spaceranger-tiny-bcl-samplesheet-1.0.0.csv.
A simple CSV sample sheet is recommended for most sequencing experiments. The simple CSV format has only three columns (Lane, Sample, Index), and is thus less prone to formatting errors. You can see an example of this in spaceranger-tiny-bcl-simple-1.0.0.csv.
Lane,Sample,Index 1,test_sample,SI-TT-D9
Listed below are the descriptions for each column.
| Column | Details |
|---|---|
| Lane | Which lane(s) of the flow cell to process. Can be either a single lane, a range (e.g., 2-4) or '*' for all lanes in the flow cell. |
| Sample | The name of the sample. This name is the prefix to all the generated FASTQs, and corresponds to the --sample argument in all downstream 10x pipelines. Sample names must conform to the Illumina bcl2fastq naming requirements. Only letters, numbers, underscores, and hyphens are allowed; no other symbols, including dots ("."), are allowed. |
| Index | The 10x sample dual-index that was used in library construction, e.g., SI-TT-D9. |
To run mkfastq with a simple layout CSV, use the --csv argument. Here is how to run mkfastq on the tiny-bcl sequencing run with the simple layout (replace the highlighted code with the path to tiny-bcl on your system):
1spaceranger mkfastq --id=tiny-bcl \ 2 --run=/PATH/TO/tiny-bcl \ 3 --csv=spaceranger-tiny-bcl-simple-1.0.0.csv
The spaceranger mkfastq pipeline can also be run with a sample sheet in the Illumina Experiment Manager (IEM) format (e.g. spaceranger-tiny-bcl-samplesheet-1.0.0.csv). An IEM sample sheet has several fields specific to running on Illumina platforms, including a [Data] section where sample and index information is specified. spaceranger mkfastq supports listing either index set names or the oligo sequences.
Do not trim adapters during demultiplexing. Leave these settings blank. Trimming adapters from reads can potentially damage the 10x barcodes and the UMIs, resulting in pipeline failure or data loss.
If you are using an Illumina sample sheet for demultiplexing with bcl2fastq, BCL Convert or our mkfastq pipeline, please remove these lines under the [Settings] section: Adapter or AdapterRead1 or AdapterRead2.
Dual-indexing example
Version 1: SI-TT-D9 refers to a 10x Genomics dual-index sample index; mkfastq auto-detects that this is a dual-index sample. In this example, only reads from lane 1 will be used. To demultiplex the given sample index across all lanes, omit the Lane column entirely.
[Data] Lane,Sample_ID,index 1,test_sample,SI-TT-D9
Version 2: The index sequences for SI-TT-D9 are specified in the two index and index2 columns.
[Data] Lane,Sample_ID,index,index2 1,test_sample,TGGTCCCAAG,ACGCCAGAGG
Sample names must conform to the Illumina bcl2fastq naming requirements. Specifically only letters, numbers, underscores, and hyphens are allowed. No other symbols, including dots ("."), are allowed.
Also note that while an authentic IEM sample sheet will contain other sections above the [Data] section, these are optional for demultiplexing. To avoid data loss from trimming, we do not recommend including adapter sequences in the [Settings] section of the sample sheet (see this article for details). For demultiplexing an existing run with spaceranger mkfastq, only the [Data] section is required.
Next, run the spaceranger mkfastq pipeline, using the --samplesheet argument (replace highlighted code with the path to tiny-bcl on your system).
1spaceranger mkfastq --id=tiny-bcl \ 2 --run=/PATH/TO/tiny-bcl \ 3 --samplesheet==spaceranger-tiny-bcl-samplesheet-1.0.0.csv
Once the spaceranger mkfastq pipeline has successfully completed, the output can be found in a new folder named with the value you provided to spaceranger mkfastq in the --id option (if not specified, defaults to the name of the flow cell):
ls -l # expected output drwxr-xr-x 4 jdoe jdoe 4096 Nov 14 12:05 tiny-bcl
The key output files can be found in outs/fastq_path, and are organized in the same manner as a conventional bcl2fastq run:
ls -l tiny-bcl/outs/fastq_path/ tiny-bcl/outs/fastq_path/ # expected output drwxr-xr-x 3 jdoe jdoe 3 Nov 14 12:26 Reports drwxr-xr-x 2 jdoe jdoe 8 Nov 14 12:26 Stats drwxr-xr-x 3 jdoe jdoe 3 Nov 14 12:26 tiny-bcl -rw-r--r-- 1 jdoe jdoe 20615106 Nov 14 12:26 Undetermined_S0_L001_I1_001.fastq.gz -rw-r--r-- 1 jdoe jdoe 51499694 Nov 14 12:26 Undetermined_S0_L001_R1_001.fastq.gz -rw-r--r-- 1 jdoe jdoe 152692701 Nov 14 12:26 Undetermined_S0_L001_R2_001.fastq.gz tree tiny-bcl/outs/fastq_path/tiny_bcl/ # expected output tiny-bcl/outs/fastq_path/tiny_bcl/ └── Sample1 ├── Sample1_S1_L001_I1_001.fastq.gz ├── Sample1_S1_L001_R1_001.fastq.gz └── Sample1_S1_L001_R2_001.fastq.gz
This example was produced with a sample sheet that included tiny-bcl as the Sample_Project, so the directory containing the sample folders is called tiny-bcl/. If a Sample_Project, was not specified, or if a simple layout CSV file was used (which does not have a Sample_Project, column), the directory containing the sample folders would be named according to the flow cell ID instead.
If you want to remove the Undetermined FASTQs from the output to save space, you can run mkfastq with the --delete-undetermined flag. To see all spaceranger mkfastq options, run spaceranger mkfastq --help.
If you encounter a crash while running spaceranger mkfastq, upload the tarball (with the extension .mri.tgz) in your output directory. Replace the example email address below with your email:
spaceranger upload your@email.edu jobid.mri.tgz
where jobid is what you input into the --id option of mkfastq (if not specified, defaults to the ID of the flow cell). This tarball contains numerous diagnostic logs that we can use for debugging.
You will receive an automated email from 10x. If not, email support@10xgenomics.com. For the fastest service, respond with the following:
- The exact
spacerangercommand line you used. - The sample sheet that you used.
- The
RunInfo.xmlandrunParameters.xmlfiles from your BCL directory. - The kind of libraries you are demultiplexing (including chemistry).