Answering Your Questions about Sample Prep Optimization for Single Cell Multiomics

Find out how you can get the most out of your Chromium Single Cell Multiome ATAC + Gene Expression experiments as 10x-perts answer key questions about sample prep optimization for multiomic studies. And watch our webinar on the topic, now available on-demand as part of our comprehensive single cell multiomics webinar series.
The epigenomic landscape and gene expression are deeply linked, interacting to drive key biological functions, such as cell differentiation, development, and disease. With Chromium Single Cell Multiome ATAC + Gene Expression, you can simultaneously profile gene expression and open chromatin from the same cell. In the past, most methods for studying epigenomics and transcriptomics required these two modalities to be assayed separately, leaving researchers to integrate data and infer relationships after the fact. Now, you can get two readouts from an individual cell, allowing you to more thoroughly characterize cell types and states and gain deeper insights into underlying gene regulatory mechanisms with confidence.
With multiomic technologies, you can enhance your understanding of the relationship between the epigenome and transcriptome while maximizing precious samples. However, the quality of your results can start to take shape before your experiment even begins. For Single Cell Multiome ATAC + Gene Expression, quality sample prep begins with thinking about how the samples will be collected and potentially stored. After that, optimizing nuclear isolation is the next critical step in successful sample prep. In a recent webinar hosted by The Scientist, 10x-perts Sharmila Chaterjee, PhD, and Zuleyma Peralta, PhD took a deep dive into the world of sample preparation for single cell multiomics, focusing on the different protocols for tissue dissociation and nuclei isolation—and how to choose the best one for your sample. They also discussed ways to optimize your methods and overcome your sample prep challenges, highlighting the importance of careful sample handling, indicating key metrics for sample prep quality analysis, and more.
You can watch the full on-demand webinar now, or keep reading for a preview of some of the highlights from the Q&A following the presentation.
Processing samples prior to the workflow
How should samples be preserved for running Single Cell Multiome ATAC + Gene Expression at a later time?
Tissue samples should be preserved in liquid nitrogen (not -80°C, ideally). Cell suspensions should be cryopreserved in liquid nitrogen using the appropriate freezing media with cryoprotectant. They should not be stored as dry pellets or at -80°C.
Importantly, nuclei samples should not be preserved. They should be used immediately.
There are a number of additional stopping points throughout the workflow where reagents and samples can be safely stored for a certain period of time before continuing the experiment. You can find more information in our Single Cell Multiome ATAC + Gene Expression User Guide.
Do I need to sort my samples? If so, how?
Sorting is recommended if you’re working with fibrous tissue or a sample with a large amount of debris that regular washing and filtration cannot remove. For example, we only recommend sorting PBMCs if the sample has a high number of granulocytes. Otherwise, they don’t need it, and even without sorting out granulocytes, we’ve gotten high-quality results from PBMC samples. If your sample does require sorting, our demonstrated protocol for nuclei isolation from complex tissues provides directions for FACS sorting and sample cleanup.
If you decide to sort your sample, it’s important to note that nuclei need to be handled with extreme care during sorting. We have some recommendations for best practices, as any additional handling can further compromise the nuclear membrane and cause leakage of nuclear content. There are also factors to consider outside of sample quality. For example, the type of dye you’re using can significantly affect your results. Some dyes like DAPI and ethidium homodimer, when used for sorting specifically, have been observed to interfere with the assay.
Optimizing nuclei isolation for single cell multiomics
I have protocols for isolating nuclei for scRNA-seq and scATAC-seq. Can I use either protocol for Single Cell Multiome ATAC + Gene Expression?
We optimize each of our protocols for their specific assay, and therefore recommend using our Single Cell Multiome ATAC + Gene Expression protocols when running this assay. Protocols developed specifically for single cell RNA-sequencing (scRNA-seq) and single cell ATAC-sequencing (scATAC-seq) may not result in the highest data quality with the Single Cell Multiome ATAC + Gene Expression assay.
Please refer to these nuclei isolation protocols for your experiments with Chromium Single Cell Multiome ATAC + Gene Expression.
How do you get a clean nuclei suspension?
A clean nuclei suspension free of debris and clumps is very important for the Single Cell Multiome ATAC + Gene Expression assay. Clumps and debris can clog your chip and lead to assay failure. Clumps are also an indication that your nuclei sample has been overlysed. Overlysed nuclei may release their RNA into the solution, thus increasing background. Debris is also a notorious carrier of ambient RNA and should be avoided.
Following one of our demonstrated protocols, for PBMCs and cell lines or for tissues, you should be able to get a clean nuclei suspension from your sample. They both walk you through the process of isolating, washing, and counting your nuclei. However, if you expect your tissue will have more debris, please start your sample prep with a demonstrated protocol that’s optimized for nuclei isolation from complex tissues. It has a built-in sorting step, which helps get rid of debris and clean up the nuclei prep.
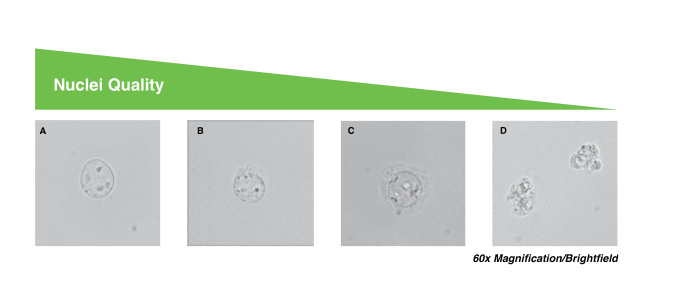
How can I evaluate the quality of my nuclei suspension?
To determine the quality of our nuclei, we generally look at membrane integrity, assessing the nuclear membranes under the microscope to see whether or not they look intact. High-quality nuclei have well-resolved edges, so we want to ensure that there is no blebbing or disintegration, where the membrane begins to form curls. You can also measure the viability of your cells using trypan blue and an automated counter or hemocytometer. Unlysed cells stain as live while nuclei stain as dead, so, if you’re lysing was successful, you should see < 5% live cells. Additionally, you can use ethidium bromide to confirm your nuclei counts, ensuring the nuclei have content and distinguishing them from debris.
Gene expression results for whole cell versus single nuclei
How does whole cell RNA-seq compare to RNA-seq from nuclei?
Nuclei have less mRNA, so fewer total number of genes and UMIs may be detected when profiling gene expression from nuclei versus whole cells. However, we have observed that cell populations recovered between whole cell and single nuclei RNA-seq are comparable, and marker expression is maintained. There are also some advantages to using single nuclei for RNA-seq. It can reduce biases in cell populations caused by cell dissociation, minimize stress-induced transcriptional artifacts, and reduce microfluidic cell capture bias due to cell size. Nuclei also enable single cell studies of intact frozen whole tissue when isolation of viable whole cells is difficult.
Ensure you get the best data from your single cell multiomics experiments with these tips for sample prep. For more details, watch our webinar on-demand, or visit our support site to explore comprehensive protocols addressing each aspect of the workflow, from sample prep to data analysis.
Looking to get started with Chromium Single Cell Multiome ATAC + Gene Expression? Explore these resources.