In addition to 10x Genomics' Cloud Analysis web application and command line (CLI) tool, you can analyze your data with conversational prompts in an AI-powered interface. A large language model (LLM) uses the 10x Cloud Model Context Protocol (MCP) server to translate your written requests into actions on the 10x Cloud app. Log in to your 10x Cloud account to verify all the results of the prompt-based interface (e.g., uploading input files, creating analyses).
Refer to the supported pipelines and products page for full details on prompt-based 10x Cloud analysis, pipeline version, and library type support. On this page, learn installation steps and example prompts for common use cases.
- Claude Desktop is a graphical user interface tool. It is a good starting point for most users to interact with the 10x Genomics Cloud using conversational prompts.
- Claude Code is an agentic tool that integrates coding and conversational prompts. It may speed up analysis for advanced users who are familiar with working in a terminal.
Please select the tab for the Claude application below to continue:
This software is required to use the prompt-based 10x Cloud interface:
- 10x Cloud account and 10x Cloud access token
- Claude Code
These data inputs are needed for analyzing data on the 10x Cloud:
- Data: FASTQ files from 10x Chromium single cell data from these products:
- Universal 3’ Gene Expression
- Universal 5’ Gene Expression
- Flex Gene Expression
- Note: The 10x Cloud accepts FASTQ sequence files from compatible sequencer platforms. Unsupported sequencing platforms may produce FASTQ file formats that are incompatible with the 10x Cloud.
- Optional: custom reference (alternatively, use pre-built human and mouse references)
- Optional: multi config CSV for
multipipeline, aggregation CSV for theaggrpipeline (alternatively, the LLM can help you create the CSV files, but may need refinement to ensure the parameters and formatting are correct)
-
Set up a 10x Cloud account: First-time users of the 10x Cloud platform must create a new account. If you already have a 10x Cloud account, there is no need to create a new one.
-
Download Claude Code: Download Claude Code following the instructions from the Anthropic website. Example shown below for installing on MacOS:
# Install Claude Code npm install -g @anthropic-ai/claude-code # Open Claude Code and follow prompts to set up and authorize account claude
-
Download 10x Genomics Cloud prompt-based interface: Install the plugins necessary for connecting to the 10x Cloud MCP. Follow the installation prompts.
# Install life-sciences /plugin marketplace add https://github.com/anthropics/life-sciences.git # Install 10x Cloud MCP /plugin install 10x-genomics@life-sciences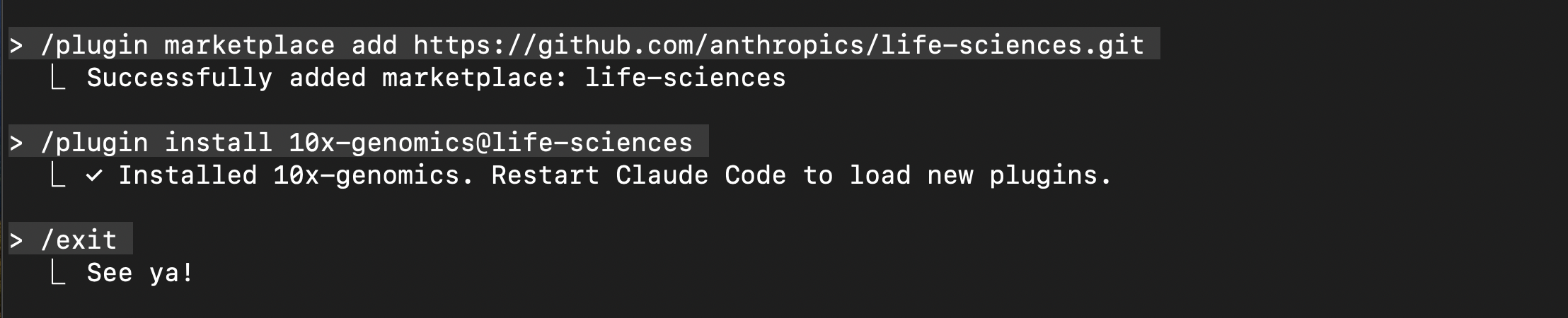
You will need to restart Claude Code after plugin installation.
# Close Claude Code exit # Restart Claude Code claudeTo complete the plugin set up, you will need your 10x Cloud Access Token, which can be found in the Security section of your 10x Cloud Account Settings.
# Complete plugin set up /plugin # Navigate with up / down keys to "Manage and uninstall plugins" > Hit Enter to open "life-sciences" > # Hit Enter to open "10x-genomics" > select "Configure" # to set up 10x Genomics Cloud MCP with 10x Cloud Access token # Follow configuration prompts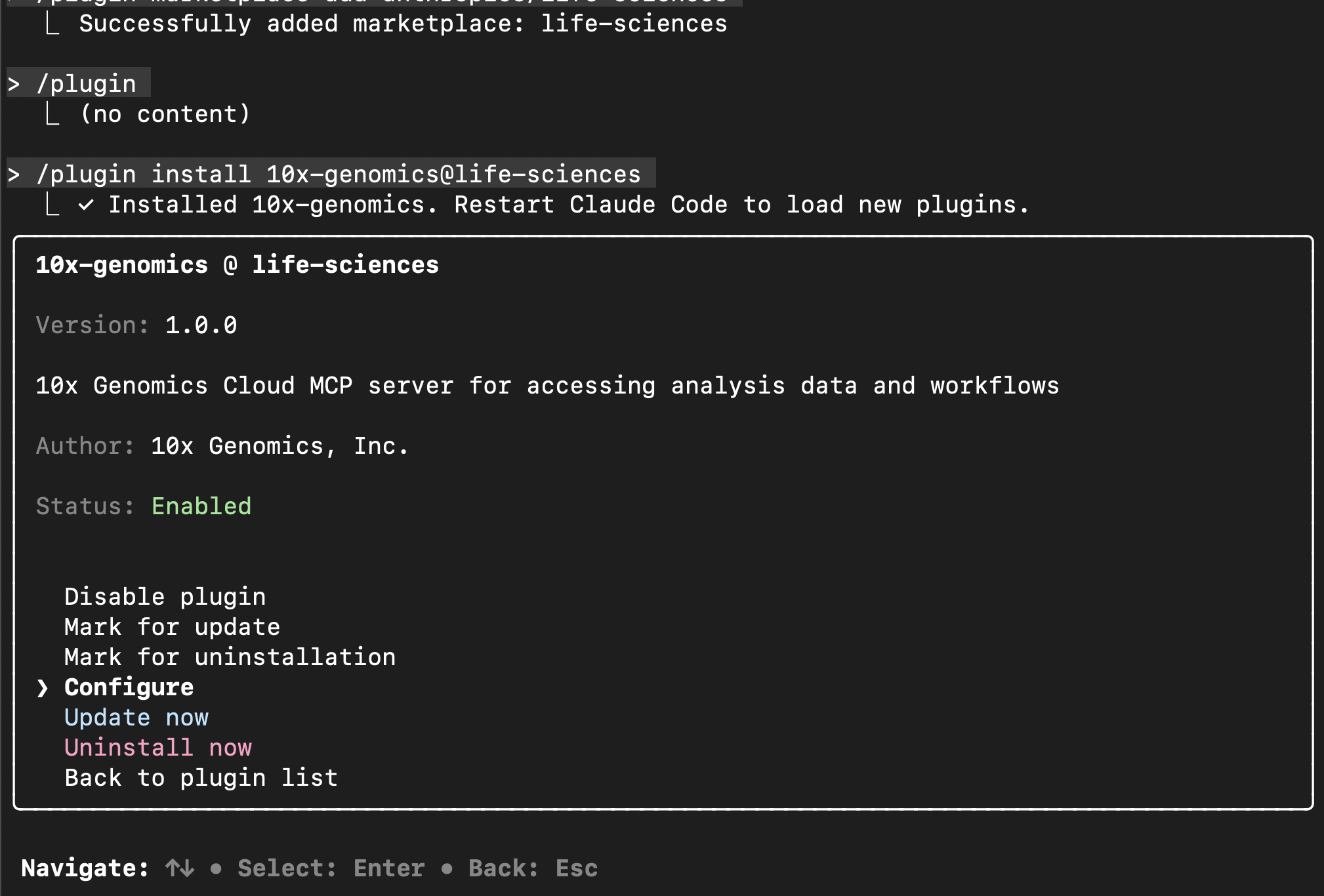
You will need to restart Claude Code after plugin configuration.
# Restart Claude Code exit claude
After verifying the MCP server is connected, you can now use both code and conversational prompts to interact with the 10x Cloud in Claude Code.
# Verify server connection
/mcp
To test the prompt-based interface, you can select from a number of single cell datasets on the 10x Genomics public datasets website and compare the prompt-based interface-created analysis results with the posted outputs on the datasets website (e.g., web_summary.html).
Before we get started with use cases, here are some tips for constructing chat prompts. It is important to be specific in your requests and queries and encourage the LLM to explain the decision-making process. The LLM can help you get started, but as with a normal conversation, it will likely require iteration to successfully create, run, and analyze a dataset.
Include specific details in your prompts about the dataset and the analysis. For example:
- Include information about the dataset species (e.g., human, mouse), sample type (e.g., cells or nuclei, disease state), and experimental setup (e.g., sample preparation, replicates, experiment conditions). These details can be useful when determining how to analyze and interpret the data.
- Include the data product family (e.g., Universal 3' Gene Expression, Universal 5' Gene Expression, Flex Gene Expression), modality (e.g., gene expression, cell multiplexing, antibody, antigen, CRISPR), and sample plexy (single vs. multiple samples). It is also helpful to indicate the library type (e.g., "GEM-X 3' Gene Expression v4"), if known.
- Include what pipeline to run (e.g.,
count,multi,aggr), as well as specific parameters, if any (e.g., create BAM files for 3' and 5' Gene Expression, but not for Flex Gene Expression analyses). Or if unsure, indicate in the prompt that you would like assistance in selecting the optimal pipeline and parameters. Theinclude_intronsparameter is set totrue(for non-Flex Gene Expression analyses) andchemistrydetection is set toautoby default.
Below, we provide example prompts and follow-up questions that you can use as templates to get started with common 10x Cloud and data analysis use cases.
The prompt-based interface has many functions, including the following:
- Create a project
- Upload input files (e.g., FASTQ, CSV)
- Create an analysis and run it on 10x Cloud
- Check analysis run status
- Download analysis output files to your computer
- List project, analysis, files, pre-build references, annotation models
- Check multi config CSV specifications
- Check billing information
Go to your Cloud Account to further manage projects and analyses. The following functions are not yet supported with the prompt-based interface:
- Cancel an in-progress analysis
- Delete an analysis
This is a good prompt to begin with following extension setup in order to confirm that Claude has access to the 10x Genomics Cloud tools:
What 10x cloud tools do you have access to?
Example follow-up prompts:
- What project settings can I update with the 10x Cloud prompt-based interface?
- List pre-built transcriptome references.
- List the annotation models available for cell annotation
- List the current analyses in all my 10x cloud projects.
- List the project files of the current project.
- List the analysis outputs from the "3prime-count" analysis.
Create a new project called: "first-mcp-project". The analysis I want to run should
be called: "3p-GEX-count". I have 1 sample of single cell Universal 3' Gene Expression
human data with 2 lanes of sequencing. The library and chemistry type is NextGEM 3'
Gene Expression v3.
The FASTQ files to upload to the Cloud are in this folder:
/Users/<user.name>/Desktop/3pGEX-count. I want to run the Cell Ranger
count pipeline.
Use the support documentation from these websites to help me set up an analysis:
https://www.10xgenomics.com/support/software/cloud-analysis/latest and
https://www.10xgenomics.com/support/software/cell-ranger/latest. Are there any
parameters for the count pipeline that I should consider for my analysis?
Example follow-up questions:
- Could you check the analysis run status?
- What is the total file size of the outputs from this analysis?
- Could you download all the outputs to my computer? Save them to my Desktop/.
In addition to creating the multi analysis, you can also learn about the configuration options directly in your chat:
How do I create a multi config csv for a cellranger multi analysis?
An example with a detailed prompt is shown below. The level of detail you provide is subjective, but more information may improve or speed up the analysis creation process. This particular example includes information provided with this 10x public dataset.
The analysis I want to run should be called: "Flex-GEX-multi", and I want
it added to an existing project called "first-claude-mcp-CR-9-flex". I have
1 sample of single nuclei Flex Gene Expression human data with 2 lanes of
sequencing. The library and chemistry type is GEM-X Flex Gene Expression.
Fresh frozen human kidney tissue was obtained from Avaden BioSciences by 10x Genomics.
The tissue was sectioned into two 44 mg samples and nuclei were isolated using
the Chromium Nuclei Isolation Kit (CG000505). A total of 4,180,000 nuclei were
obtained, which were aliquoted into a sample containing approximately 2 million nuclei.
The sample was fixed for one hour at room temperature using the Fixation of Cells & Nuclei
for GEM-X Flex Gene Expression protocol (CG000782). Following fixation, probe
hybridization was performed according to the user guide. 300,000 nuclei aliquots were
used for hybridization. After hybridization, the samples were washed and filtered according
to the protocol. The sample was then resuspended, counted, and loaded into a
Chromium GEM-X Chip. 4,000 nuclei were targeted. Gene Expression libraries for GEM-X Flex
were generated using the GEM-X Flex Gene Expression Reagent Kit for Singleplex samples
(CG000786). Libraries were sequenced on an Illumina NovaSeq 6000, with a mean read
depth of approximately 20,000 reads per cell using a paired-end, dual indexing
sequencing scheme.
The FASTQ files are already uploaded to this project: "first-claude-mcp-CR-9-flex".
I want to run the Cell Ranger multi pipeline. Use the support documentation from these
websites to help me set up an analysis:
https://www.10xgenomics.com/support/software/cloud-analysis/latest and
https://www.10xgenomics.com/support/software/cell-ranger/latest. Are there any
parameters for the multi pipeline that I should consider for my analysis?
Below is an example of setting up a more complicated Cell Ranger multi analysis, in addition to iterating on the analysis with the same inputs but changing some parameters.
I want to set up another cellranger multi analysis in the "first-claude-mcp-CR-9-flex" project.
The analysis should be called "3pgex-cmo-crispr-multi". This dataset is Universal 3' Gene
Expression v3 data with a Cellplex (Cell Multiplexing) library and a CRISPR library.
There is 1 sample, and 3 feature types: Gene Expression, Multiplexing Capture, and CRISPR
Guide Capture. The CellPlex experiment uses 4 CMO tags (CMO308, CMO309, CMO310, and CMO311).
This is a human sample, so I want to use a pre-built human transcriptome reference.
The FASTQ files for all these libraries are located in:
/Users/<user.name>/Desktop/3pgex-crispr-cmo. The feature reference is in the same
path, called "feature_ref.csv". What analysis considerations or parameters should I
think about before running this analysis?
Note that analysis iteration within Claude should work on analyses created by the MCP, but not analyses created by other methods (e.g., directly in the Cloud web app or TXG CLI).
Can you start a new run with the same 3 libraries (GEX, CMO, and CRISPR), but this time
treat it as 1 sample pooled from 4 tubes. In this case, the samples part of the config CSV
should look like this:
[samples] sample_id,cmo_ids,description gex_crispr_cmo_4tags,CMO308|CMO309|CMO310|CMO311,4tags
Example prompts:
- Can you explain what the R1, R2, I1, and I2 FASTQ files are?
Provide the resources used to compile this information.
- What is the difference between the Cell Ranger count and multi pipelines?
Which is best for Flex Gene Expression data analysis? Provide the resources
used to compile this information.
- What are the main pipeline analysis steps used to analyze my data with
the count pipeline? Provide the resources used to compile this information.
Tip: Set up the Filesystem Desktop Extension for this use case, as it makes it easier for Claude to find the files.
I have many samples of data on my computer at /Users/<path>/<directory>.
Can you upload all of the FASTQ files into a new project and start Cell Ranger count
analyses for all of them at the same time? Infer the correct Cell Ranger analyses to create
based on the sample names, and ask for my confirmation before creating the analyses.
Claude Desktop cannot currently read downloaded HTML files > 1MB (Claude Code is able to parse larger files). However, you can ask the LLM to read other output CSV files or type metric results into the chat and ask for interpretation guidance.
- Read the metrics_summary.csv file and explain the results.
- Read the cell_types.csv file. Which are the most common cell types in my dataset?
If you have feedback about this feature or need help troubleshooting, reach out to support@10xgenomics.com.