Data integration is the process of merging multiple experiments into a single dataset in order to boost statistical power and enable robust biological comparisons. An important aspect of this process is eliminating confounding technical variations.
Cloud Analysis offers two workflows for integrating data generated from different samples:
- Normalization (SCTransform) and batch correction (Harmony) workflow
- Data aggregation with Cell Ranger aggr pipeline
The optimal choice depends on your experimental design and analysis objectives. Key differences and guidance on when to use each workflow are detailed in the FAQs section.
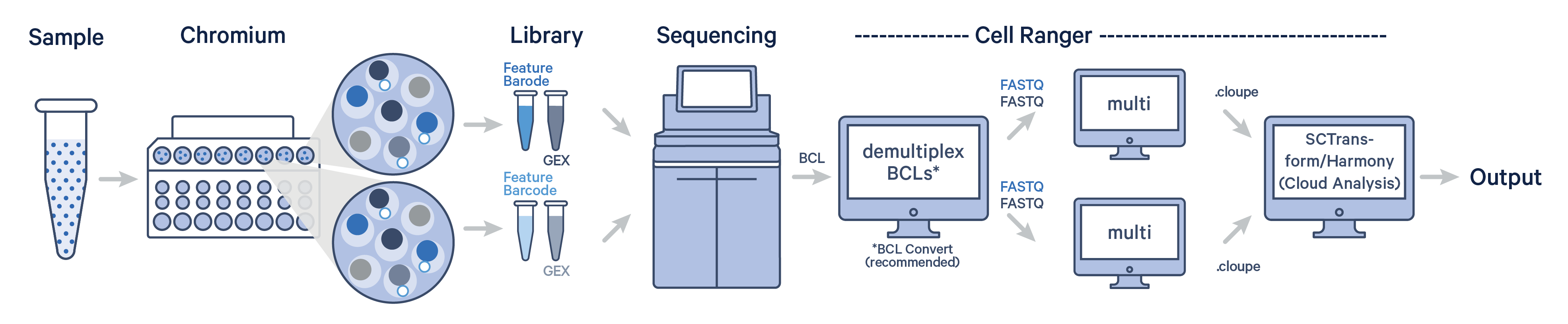
The industry-standard SCTransform/Harmony workflow for single cell data normalization, variance stabilization, and batch correction is now available on the 10x Genomics Cloud Analysis platform.
SCTransform normalizes Gene Expression data within each sample, accounting for technical variation and stabilizing variance, whereas Harmony integrates data from multiple samples, removing batch-specific effects while preserving biological differences. The goal of this workflow is to transform the raw UMI counts within a single dataset to account for technical variation and prepare the data for downstream analyses like dimensionality reduction and clustering.
The SCTransform/Harmony workflow is only available on Cloud Analysis. The workflow supports standard Cell Ranger count and multi outputs, enabling the integration of Gene Expression (GEX) data from Universal 3', 5', and Flex chemistries.
Here, you will learn how to configure the analysis and understand the output files generated.
Input requirements:
- Requires Cell Ranger
.cloupefiles output by thecellranger countorcellranger multipipeline. See list of permitted libraries and assay chemistries. - The
.cloupefiles can be generated on Cloud Analysis or manually uploaded. - Each dataset must contain a Gene Expression library.
- If VDJ or Feature Barcode (Antibody Capture or CRISPR Guide Capture) libraries are present, they will be ignored for normalization and batch correction purposes.
Cell limit:
- The analysis supports the integration of up to 500,000 cells.
.cloupe file creates new UMAP and t-SNE projections that will not include any batch correction applied during integration. Learn how to perform reanalysis in Loupe Browser.The SCTransform/Harmony workflow uses Seurat's SCTransform for normalization and Harmony for batch correction. These popular R-based tools are widely recognized in the scientific community and are supported by benchmarking studies. The specific versions are:
- R: 4.4.1
- SCTransform: 0.4.1
- Harmony: 1.2.1
- Seurat: 5.1.0
The data integration workflow does not check for feature inconsistencies between .cloupe files being merged. These inconsistencies can include, but are not limited to, differences in transcriptome references, different number of features (genes), and whether or not introns were included during data generation. Users should have a thorough understanding of how their individual .cloupe files were generated before integration. Learn how the pipeline handles .cloupe files with inconsistent feature sets.
This analysis is designed to be compatible with data from any species. For human and mouse, automated cell type annotation will be performed on the integrated dataset using the default species-specific annotation model.
Integrating datasets from different species will likely generate an error due to the lack of shared features.
Limitations
.cloupefiles aggregated usingcellranger aggrare not supported- Previously batch-corrected
.cloupedata cannot be integrated again raw_cloupe.cloupefiles from the Cell Rangermultipipeline are not supported
Step 1: Uploading .cloupe files
- Ensure your
.cloupefiles are readily available. If you need to perform initial data exploration or reanalysis, use Loupe Browser. See Filtering and Reanalysis Workflow in Loupe Browser for detailed instructions. - In your project folder, click the upload files button and upload your prepared
.cloupefiles. Alternatively, select.cloupes from a prior Cloud Analysis run ofcellranger countorcellranger multi.
Step 2: Initiating analysis
- If you uploaded
.cloupe files, check the box next to each file you wish to include in the analysis. Then Click the 'Create New Analysis' button.
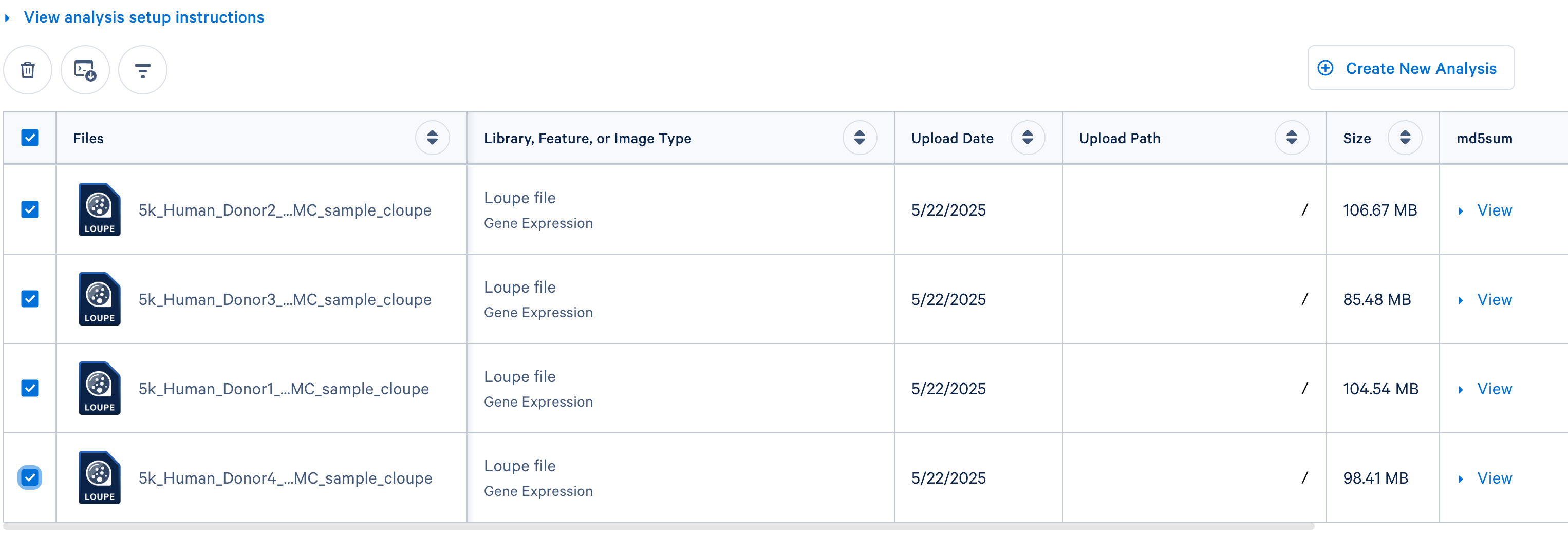
- If you are integrating
.cloupefiles from an existing Cloud Analysis run ofcountormulti, click the 'Integrate Multiple Analyses' button in the 'Analysis' tab. Then select the SCTransform/Harmony option:
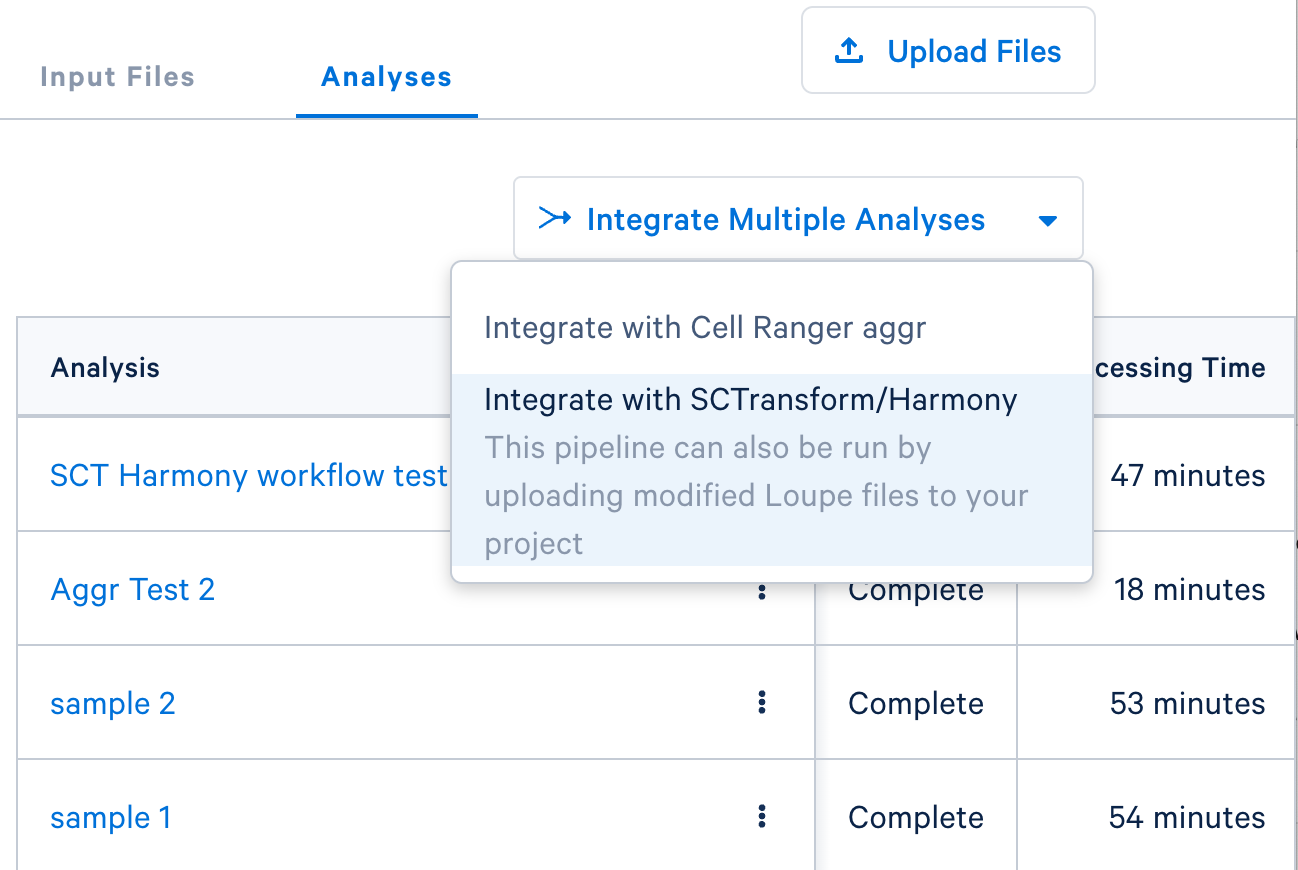
- Choose the
.cloupefiles you wish to integrate and click 'Continue' to configure the analysis.
Step 3: Configure Dataset Integration Settings
- Analysis details: Provide a descriptive name for your analysis. This will help you identify it later. Add a brief description (optional) outlining the purpose and key features of your analysis.
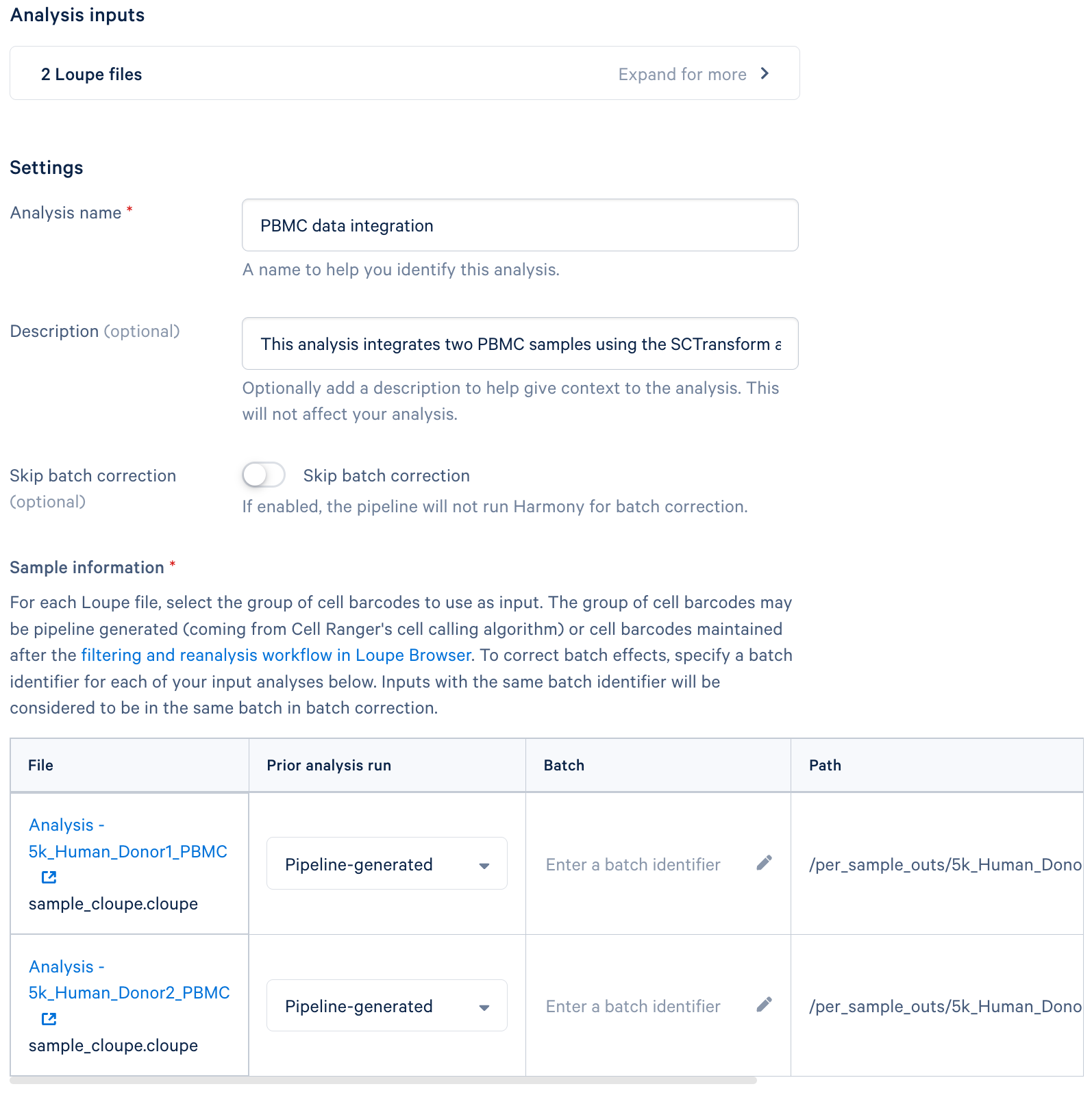
- Integration Options:
- Skip batch correction (optional): Select this option if you do not want to perform batch correction. Typically, batch correction is necessary for removing technical variations between samples.
- While this workflow facilitates the merging of Gene Expression data across different experimental setups, only Gene Expression libraries are integrated. Any additional libraries like VDJ, Antibody Capture, and CRISPR are ignored and the data from these libraries will not be included in the output. See the FAQ section Which libraries are permitted? for details.
- Sample information:
- This section is for specifying how batch correction should be performed. You may choose to select the original pipeline-generated cells or cells generated after performing Loupe Browser's Reanalyze workflow.
- Batch: Assign a unique identifier (e.g., "Treatment1", "Treatment2") to each file. Files with the same batch identifier will be grouped together for batch correction.
Step 4: Understanding advanced options
The "Advanced Options" section allows you to fine-tune the SCTransform parameters. These options are useful for optimizing performance based on your specific dataset characteristics.
- Number of cells:
- This parameter controls the number of cells randomly sampled from the dataset to estimate SCTransform model parameters. A representative subset is used to speed up processing while preserving model accuracy.
- When to use: Lowering this value reduces computation time but may reduce normalization quality if your dataset has high diversity.
- Variable features:
- This option determines the number of top variable genes (features) to keep after variance modeling in SCTransform. These are used for downstream steps like PCA and clustering.
- When to use: Increasing this number captures more features but may introduce noise; decreasing focuses on the most informative genes, reducing noise and dimensionality, but risking loss of subtle variations.
- Only variable genes:
- If enabled, only variable genes are retained in the SCTransform normalized data. Nonvariable genes are excluded to save memory and focus analysis.
- When to use: This can significantly reduce the size of the output data, improving computational efficiency and focusing on the most informative genes. If you have a very large gene set, and you know that most of the variation is held within a smaller subset of genes, this will be very helpful.
Step 5: Running the analysis
- Once you have configured all the settings, click the 'Run Analysis' button. You will receive an email notification when your analysis is complete.
| File | Description |
|---|---|
| Integrated Loupe Browser file | A single Loupe Browser file (.cloupe) containing raw count data and integrated secondary analysis outputs from all input Loupe Browser files. It enables interactive visualization and exploration of the combined dataset within the Loupe Browser. |
| Integrated Feature/Cell matrix | A feature-barcode matrix in HDF5 format, storing the raw counts for all cells across all input datasets. |
| Integrated Seurat object | A Seurat object saved in the .qs format, offering efficient storage and quick uploading for downstream analysis in R. Loading this object requires the qs R package. Learn how to read Seurat object in R |
| Cell annotation report | A summary of the cell types found in the sample. |
| Cell annotation details | Data files containing more detailed information about cell type assignments. |
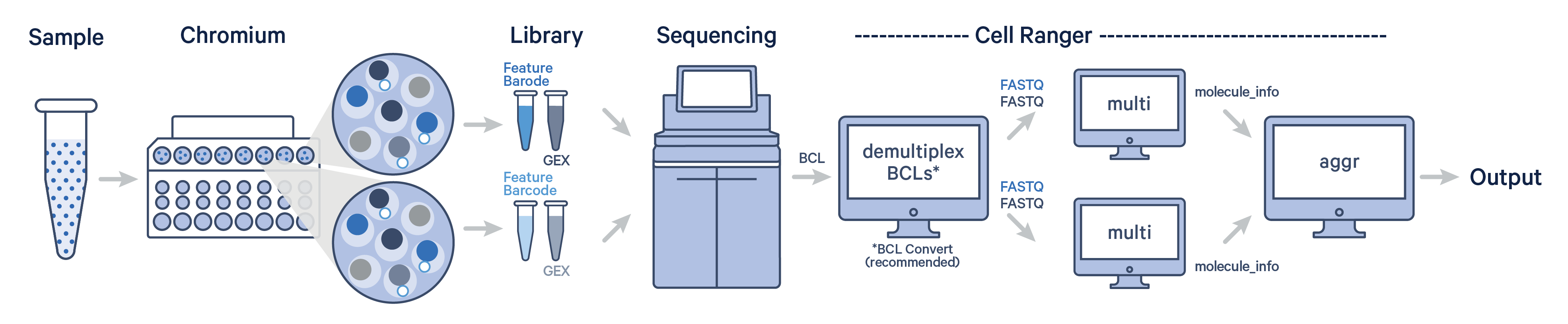
The Cell Ranger aggr pipeline can be used for combining the output files (molecule_info.h5) from multiple independent runs of Cell Ranger count, multi, or vdj. It is a data aggregation tool, part of the 10x Genomics suite.
Cell Ranger aggr generates a unified feature-barcode matrix and resolves potential barcode collisions by adding run-specific suffixes. Importantly, cellranger aggr uses a distinct normalization and chemistry batch correction method compared to SCTransform and Harmony. See FAQ section What is the difference between aggr and SCTransform/Harmony workflow for details.
A single GEM well can yield multiple physical libraries: one Gene Expression library and one or more Feature Barcode libraries. The example below has one Gene Expression library per GEM well.
Here, each Gene Expression library was analyzed as separate runs of the Cell Ranger multi pipeline. Select the FASTQ files for a given library, click 'Create New Analysis'.
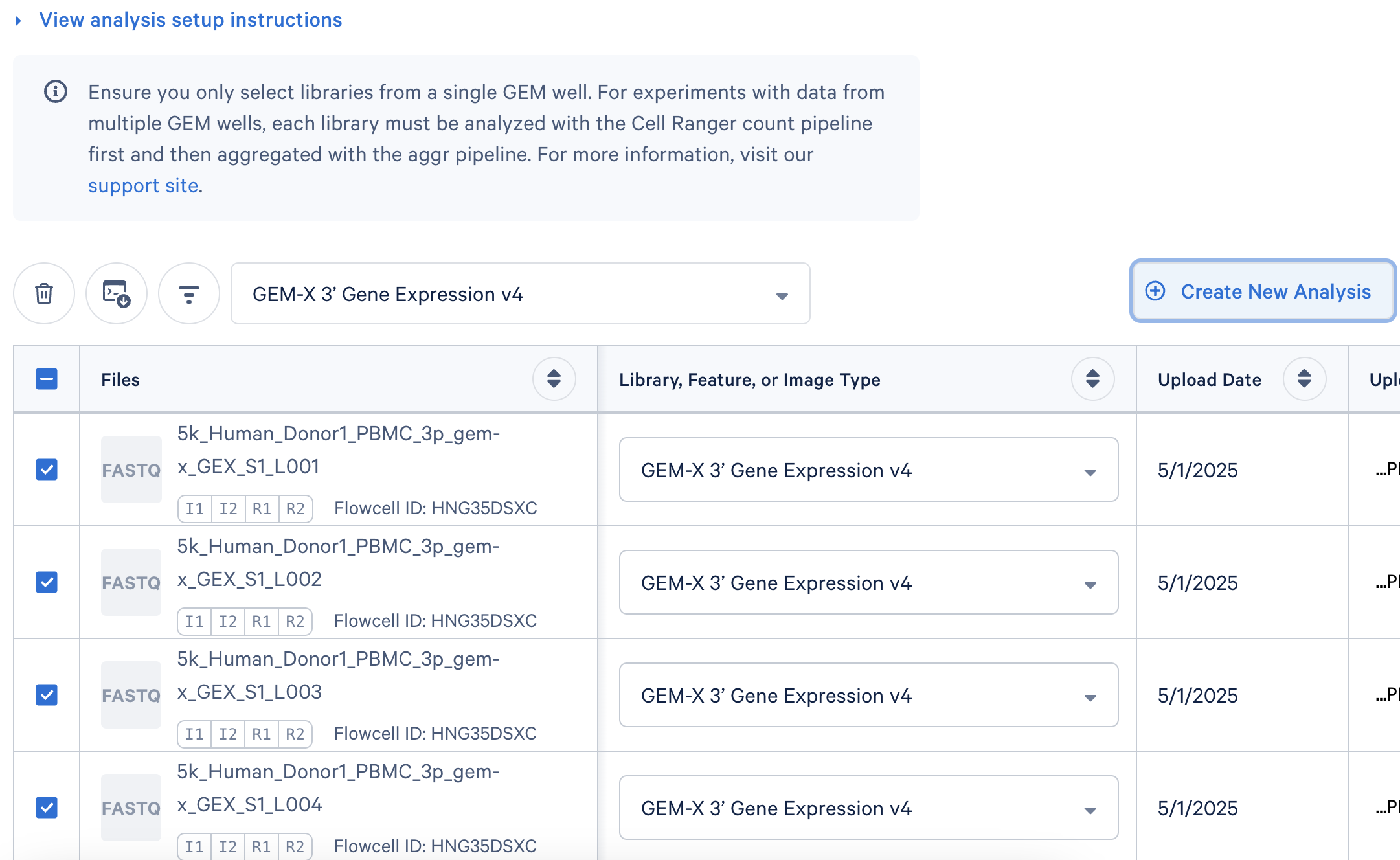
The Upload Path is blank for files uploaded with the Web Browser 'Browse Files' option.
Once the multi analyses are complete for each library, the aggregation option will be visible:
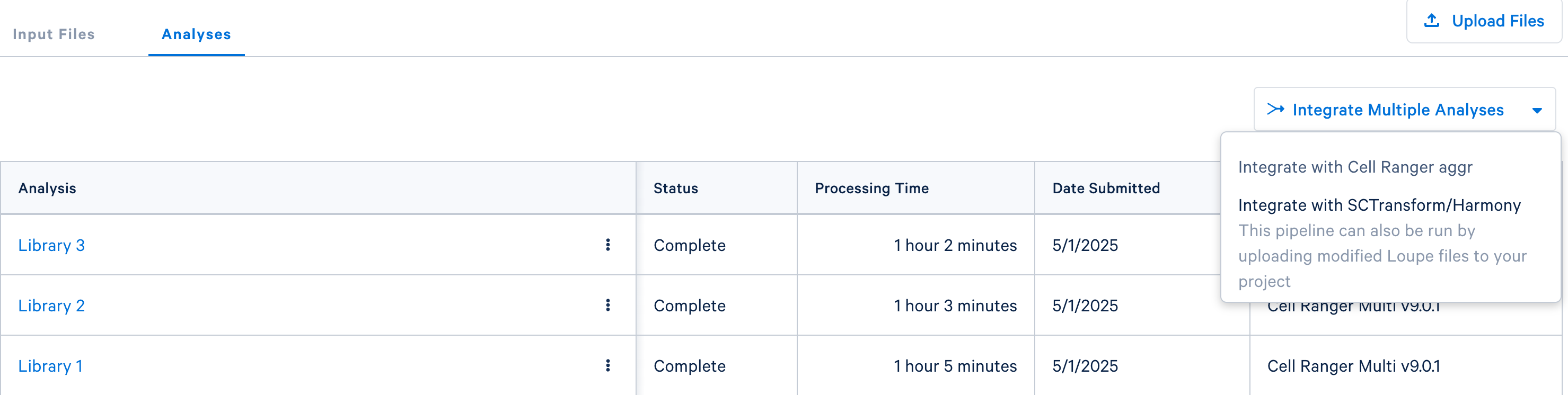
Click the 'Integrate with Cell Ranger aggr' button to choose the datasets to aggregate and fill out the set up form to run the analysis. The 10x Cloud Analysis set up form autogenerates the aggregation CSV file; users do not need to create one. This file will be available for inspection and download after the analysis has been created.
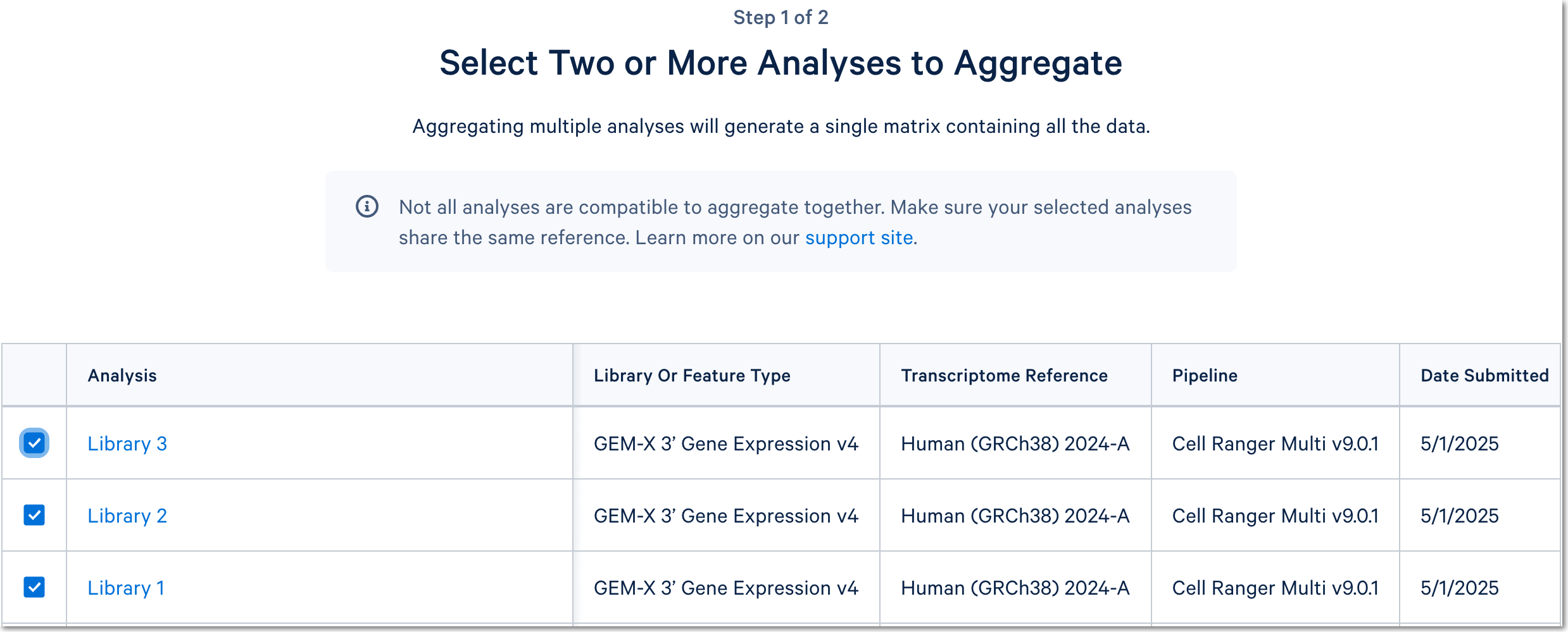
For a more detailed guide, see this Getting Started tutorial for Cell Ranger aggr. The tutorial was written for the command line version of the aggr pipeline.
The Cell Ranger Aggregate Outputs page describes the outputs that are generated after a successful aggr run.
The aggr pipeline, part of Cell Ranger, is specifically designed to mitigate batch effects when aggregating data from multiple Cell Ranger runs with different chemistries. The SCTransform/Harmony workflow offers an alternative strategy using popular R packages for normalization and batch correction, providing broader options for combining datasets compared to Cell Ranger's aggr.
The following table provides a comparison of both workflows:
| Comparison Point | Cell Ranger aggr | SCTransform/Harmony |
|---|---|---|
| Normalization | Equalizes the average read depth per cell between groups before merging | SCTransform (R tool) performs normalization using regularized negative binomial regression, modeling the mean-variance relationships. |
| Batch Correction | MNN (Mutual Nearest Neighbors) | Harmony (R tool) is run on the normalized data to remove batch effects across multiple samples. |
| Input | molecule_info.h5 files from Cell Ranger runs | .cloupe files |
| Environment | Can be run on local environment or on Cloud Analysis | Cloud Analysis only |
| Origin | Part of the standard Cell Ranger suite | Relies on third-party R tools |
| Flexibility | Primarily focused on chemistry batch correction | Normalization and batch correction within and across chemistries |
| Usage Constraints | Not suitable for combining datasets from filtered .cloupe files. | Cloud Analysis-only workflow. Do not use if your data have Cloud Analysis restrictions. |
| Pipeline Compatibility | Cell Ranger, Cell Ranger ARC | Cell Ranger |
Integration with SCTranscform/Harmony
- Existing Cloud Analysis: You can use
.cloupefiles generated from previouscount/multianalyses performed within Cloud Analysis. - Cell Ranger command line: If you ran Cell Ranger locally, you can upload the resulting
.cloupefiles to Cloud Analysis for integration. - Reanalyzed output: If you perform a reanalysis using Cell Ranger or Loupe Browser, you can save a
.cloupefile containing filtered barcodes, which can then be used for integration. - LoupeR:
.cloupefiles generated using the LoupeR R package are not compatible for integration.
Integration with Cell Ranger aggr
- You can only use output files generated from previous analysis performed within Cloud Analysis.
Integration with SCTranscform/Harmony
| Library/Assay | Acceptance Status (with Rejection Stage) | Notes |
|---|---|---|
| Gene Expression Only | Accepted | Processes Gene Expression data from a combination of 3', 5', and Flex libraries. |
| Gene Expression + Feature Barcode (Antibody Capture, CRISPR) | Accepted | Processes Gene Expression data; FB data is currently ignored by the workflow. |
| Gene Expression + ATAC (Multiome) | Rejected (Upload) | Multiome is not supported. To include GEX data from Multiome libraries in an integration analysis, first process the GEX portion using Cell Ranger (without the ATAC data). You can then integrate the resulting .cloupe files. |
| Antibody Capture only (no GEX) | Rejected (Preflight) | Antibody Capture without GEX is not supported |
| ATAC Only | Rejected (Upload) | ATAC-only data is not supported. |
| Visium | Rejected (Upload) | Visium data is not supported. |
Previously Aggregated with aggr | Rejected (Preflight) | Aggregated .cloupe files are not supported. Please upload .cloupe files from individual runs. |
Raw .cloupe from multi (Gene Expression-only) | Accepted | Although the workflow allows upload of raw .cloupe files, integrating them is not recommended and not supported. Integration analysis must be performed on sample-specific .cloupe files. |
Integration with SCTranscform/Harmony
If the .cloupe files have inconsistencies such as differing transcriptome versions, species, assay chemistries (e.g., Universal 3' vs. Flex), or the presence of unique custom genes, the SCTransform/Harmony workflow identifies features shared across all input files. It then performs the integration analysis using only these common features.
If no common features are found across all .cloupe files, the integration analysis will result in a preflight error.
Integration with SCTranscform/Harmony
The SCTransform/Harmony integration workflow allows a single .cloupe file to be processed up to five times. However, once a .cloupe file has been batch-corrected, it is no longer a valid input for subsequent batch correction workflows.
Integration with SCTranscform/Harmony
- All output files, except for the Seurat object, are free forever.
Integration with aggr
- All output files are free forever.
Integration with SCTranscform/Harmony
No, but there is a limit on the number of cells. You can integrate a maximum of 500,000 cells. Individual .cloupe files should have no more than 320,000 cells.
Integration with SCTranscform/Harmony
You can integrate a maximum of 500,000 cells.