Choose a product below to filter the page content to your needs:
Cell Ranger v7.0 and later supports analyzing Fixed RNA Profiling (FRP) data with the cellranger multi pipeline (see library compatibility table). FRP data cannot be analyzed with the cellranger count pipeline.
First, generate FASTQ files. For example, if the flow cell ID was HAWT7ADXX and you use cellranger mkfastq to demultiplex, the output FASTQ files will be in HAWT7ADXX/outs/fastq_path.
If you are already starting with FASTQ files, you can skip this step and proceed directly to run cellranger multi.
Running cellranger multi requires a configuration ("config") CSV, described below, and the following arguments:
| Argument | Description |
|---|---|
--id | A unique run ID string: e.g. sample345 that is also the output folder name. Cannot be more than 64 characters. |
--csv | Path to config CSV file with input libraries and analysis parameters. |
The multi config CSV contains both the library definitions and experimental design variables. The required sections differ slightly for analysis with single-sample ("singleplex") vs. multiplexed configurations. It is composed of up to four sections for FRP data:
- The
[gene-expression]section has two columns that specify parameters relevant to analysis of gene expression data, such as reference genome and cell-calling parameters that apply to the whole library in the case of singleplex Fixed RNA Profiling. - The
[feature]section has two columns that specify parameters relevant to analysis of Feature Barcode libraries; required when analyzing singleplex or multiplex Fixed RNA Profiling data with Cell Surface Protein ("Antibody Capture" library). - The
[libraries]section has three required columns that specify where the input FASTQ files may be found. - The
[samples]section has two required columns that specify sample information for Fixed RNA Gene Expression with multiple samples or a single sample with multiple Probe Barcodes; it is not valid for singleplex Fixed RNA Profiling analysis.
Go to the Cell Ranger Multi Config CSV page for a complete list of options for each section.
Generate a multi config CSV template by running cellranger multi-template, see usage here.
Example formats for different product configurations are illustrated below, or download examples from Single Cell Gene Expression Flex public datasets here.
After determining the input arguments and config CSV parameters, run cellranger multi (replace example code with your file path and ID name):
cd /home/jdoe/runs
cellranger multi --id=sample345 --csv=/home/jdoe/sample345.csv
Following a series of checks to validate input arguments, the cellranger multi pipeline stages will begin to run:
Martian Runtime - v4.0.8
Running preflight checks (please wait)...
...
By default, Cell Ranger will use all of the cores available on your system to execute pipeline stages. You can specify a different number of cores to use with the --localcores option; for example, --localcores=16 will limit Cell Ranger to using up to sixteen cores at once. Similarly, --localmem will restrict the amount of memory (in GB) used by Cell Ranger.
The pipeline will create a new folder named with the run ID you specified using the --id argument (e.g. /home/jdoe/runs/sample345) for its output. If this folder already exists, Cell Ranger will assume it is an existing pipestance and attempt to resume running it.
A successful cellranger multi run should conclude with a message similar to this:
Waiting 6 seconds for UI to do final refresh.
Pipestance completed successfully!
yyyy-mm-dd hh:mm:ss Shutting down.
Saving pipestance info to "tiny/tiny.mri.tgz"
The pipeline outputs will be saved in a folder named with the run ID you specified (e.g. sample345). The subfolder named outs/ contains the pipeline outputs.
Here are a few example multi config CSVs for some common Fixed RNA Profiling (FRP) assay configurations, along with simplified diagrams for the corresponding experimental set up. Make sure to replace the example path with the actual absolute path to your data and edit any text according to the experiment's sample/library/file names.
Important: In the examples below, we set no-bam to "true" so Cell Ranger will not generate a BAM file. This setting is recommended for Fixed RNA Profiling libraries and will reduce both the total computation time for the pipestance and the size of the output directory.
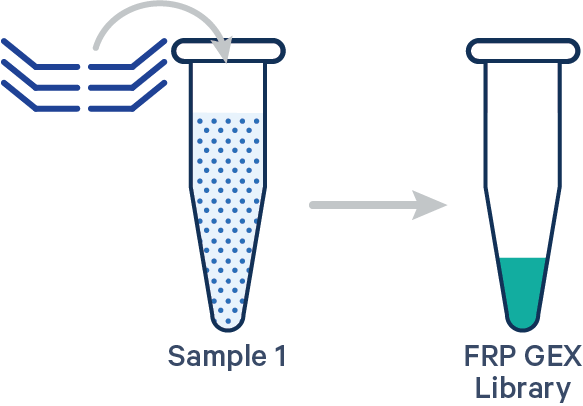
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
no-bam,true #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
frp_gex,/path/to/fastqs,Gene Expression
This library configuration does not use the [samples] section in the multi config CSV. The sample ID will be specified by the cellranger multi --id input. See example dataset.
Antibody Capture is compatible in this configuration. There is one sample, one Probe Barcode, and two libraries (Gene Expression and Antibody Capture).
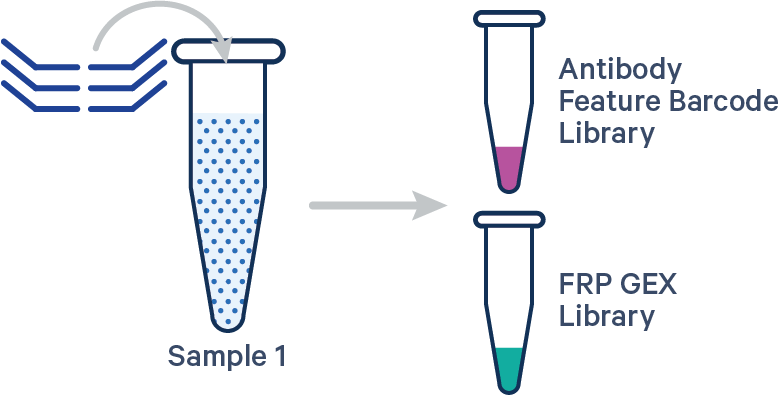
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
no-bam,true #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
frp_gex,/path/to/fastqs,Gene Expression
frp_ab,/path/to/fastqs,Antibody Capture
[feature]
reference,/path/to/feature_reference.csv
This library configuration does not use the [samples] section in the multi config CSV. The sample ID will be specified by the cellranger multi --id input. See example dataset.
Multiple biological samples
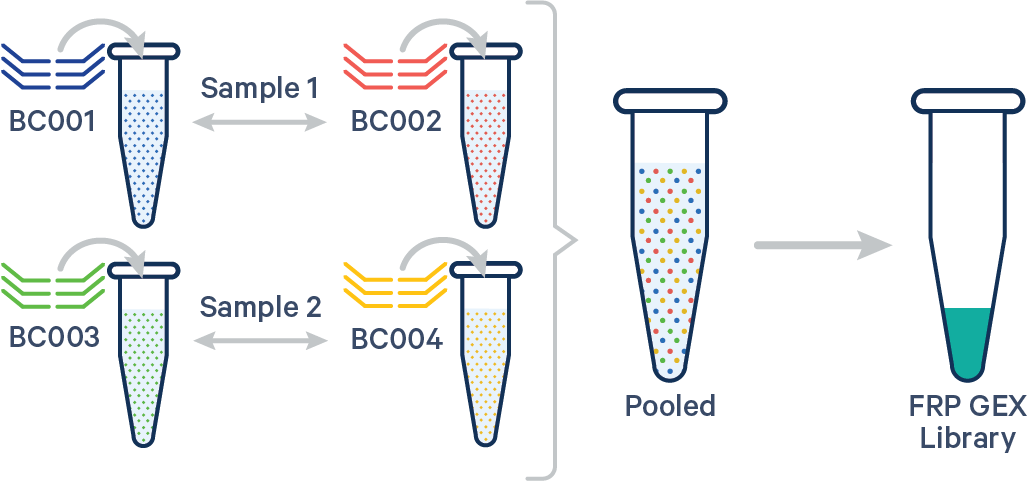
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
no-bam,true #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
frp_gex,/path/to/fastqs,Gene Expression
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001|BC002,Control
sample2,BC003|BC004,Treated
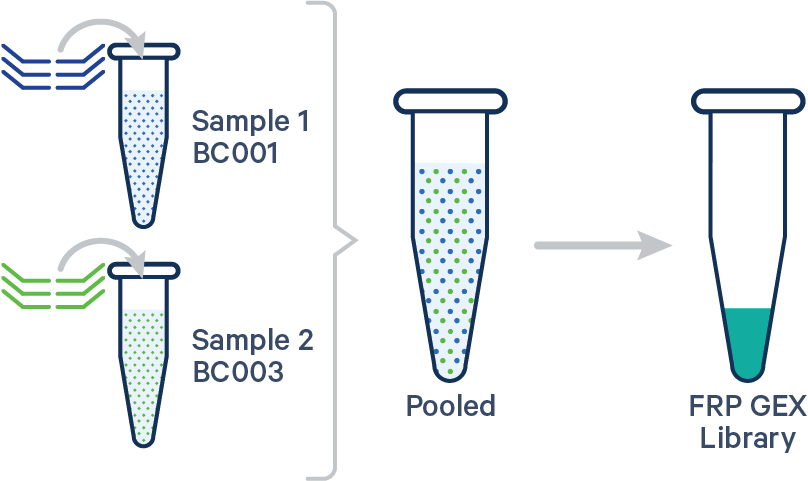
Single biological sample
In this case, the config CSV must include a [samples] section to specify the Probe Barcodes since two Probe Barcodes were used for a single sample in this experiment.
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
no-bam,true #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
frp_gex,/path/to/fastqs,Gene Expression
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001|BC002,Control
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
no-bam,true #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
frp_gex,/path/to/fastqs,Gene Expression
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001,Control
sample2,BC003,Treated
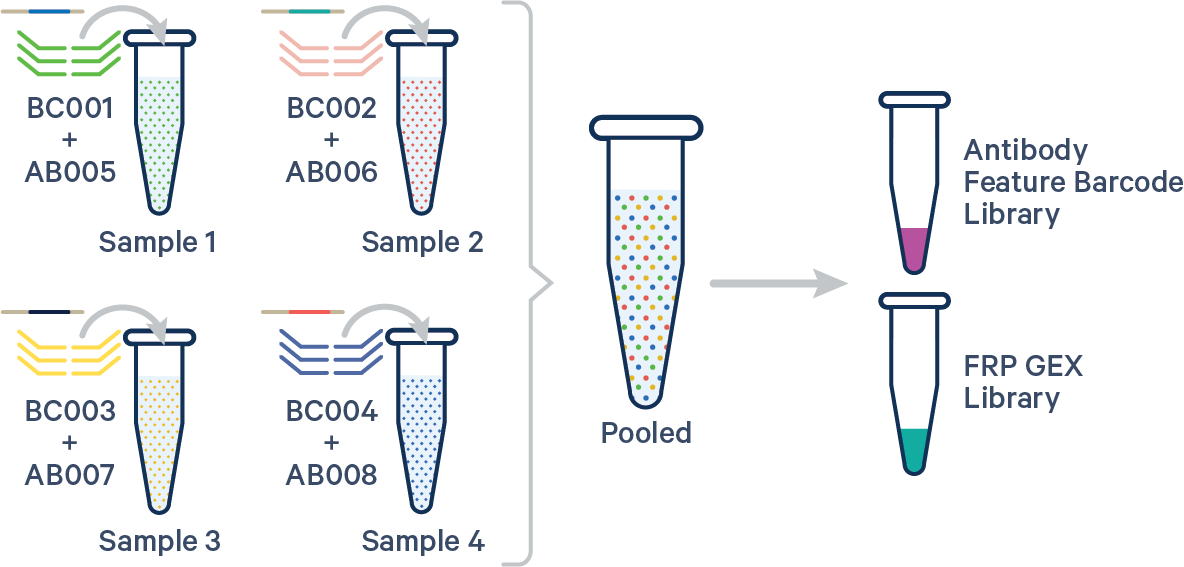
(Recommended) Specify both the Probe Barcode and Antibody Multiplexing Barcode pairs:
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
no-bam,true #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
frp_gex,/path/to/fastqs,Gene Expression
frp_ab,/path/to/fastqs,Antibody Capture
[feature]
reference,/path/to/feature_reference.csv
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001+AB005
sample2,BC002+AB006
sample3,BC003+AB007
sample4,BC004+AB008
(Advanced) Specify the Probe Barcode only, Antibody Multiplexing Barcode auto-detected:
[gene-expression]
reference,/path/to/transcriptome
probe-set,/path/to/probe-set.csv #e.g., cellranger-x.y.z/probe_sets/Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
no-bam,true #do not generate BAM file
[libraries]
fastq_id,fastqs,feature_types
frp_gex,/path/to/fastqs,Gene Expression
frp_ab,/path/to/fastqs,Antibody Capture
[feature]
reference,/path/to/feature_reference.csv
[samples]
sample_id,probe_barcode_ids,description
sample1,BC001
sample2,BC002
sample3,BC003
sample4,BC004
The v1.0.1 probe set reference CSV files for human and mouse can be found in the probe_sets directory of the Cell Ranger package (v7.1 and later):
cellranger-7.2.0/probe_sets/
└── Chromium_Human_Transcriptome_Probe_Set_v1.0.1_GRCh38-2020-A.csv
└── Chromium_Mouse_Transcriptome_Probe_Set_v1.0.1_mm10-2020-A.csv
The v1.0.1 and v1.0 probe set reference CSV files and additional support files for human and mouse:
- Can be downloaded from the Cell Ranger Downloads page
- Are described in detail in Probe Sets Overview
The following Gene Expression (GEX) assay chemistry combinations may be aggregated with cellranger aggr, and are either supported (validated by 10x Genomics) or possible (will run, but not the validated intended usage).
Some assay chemistry combinations are supported for aggregation as long as they have the same feature reference CSV file (+) and/or probe set reference CSV file (^); please see this FAQ page for details about aggregating these assay combinations.
| Assay chemistry | Singleplex FRP | Singleplex FRP, Antibody (TotalSeq-C) | Multiplex FRP | Multiplex FRP, Antibody |
|---|---|---|---|---|
| Singleplex FRP | Supported^ | - | - | - |
| Singleplex FRP, Antibody (TotalSeq-C) | Supported+^ | Supported+^ | - | - |
| Multiplex FRP | Supported^ | Supported+^ | Supported^ | - |
| Multiplex FRP, Antibody | Supported+ | Supported+ | Supported+ | Supported+^ |
| 3' or 5' GEX library | Possible | Possible+^ | Possible | Possible+^ |
The v7.2 cellranger aggr pipeline normalizes Fixed RNA Profiling sample reads by downsampling the usable reads.
We recommend rerunning v7.0 - 7.1 dataset(s) with the v7.2 cellranger multi pipeline to aggregate FRP data. The earlier versions (v7.0 - 7.1) normalized reads by downsampling the mapped reads in cells rather than usable reads due to an error in how usable reads were recorded in the molecule_info.h5 file; this has been fixed in v7.2.