Genes that resist: Understanding cancer immune evasion with single cell CRISPR screens

Complex molecular circuits enable cancer cells to resist the effects of immune checkpoint inhibitors (ICIs), molecules intended to unleash T cells to destroy cancer. To tease out the details of these evasion tactics, scientists are turning to multimodal single cell CRISPR screens, which enable functional characterization of participating regulatory drivers and their molecular pathways at scale, to investigate previously hidden mechanisms of evasion that may pose novel therapeutic targets. Learn about recent advances in this field with two studies out of the New York Genome Center, NYU, and the Broad Institute of MIT and Harvard.

Understanding how cancer cells resist immunotherapy
Despite the clinical successes of T-cell mediated cancer immunotherapy, various biological hurdles prevent it from achieving broad efficacy across patients and cancer types. One such hurdle is presented by cancer cells themselves. Keen survivalists, cancer cells are known to increase expression levels of PD-L1, a surface molecule targeted by immune checkpoint inhibitor molecules, such as the anti-PD-L1 antibodies depicted below. When left unblocked, PD-L1 can interact with the programmed cell death (PD)-1 receptor on T cells to inhibit their activation and thus suppress their killing function (1). Understanding how cancer cells regulate PD-L1 expression and discovering potentially hidden, alternative mechanisms of immunotherapy evasion are important areas of research with the potential to identify new therapeutic targets that can, in turn, improve therapeutic efficacy and patient outcomes.
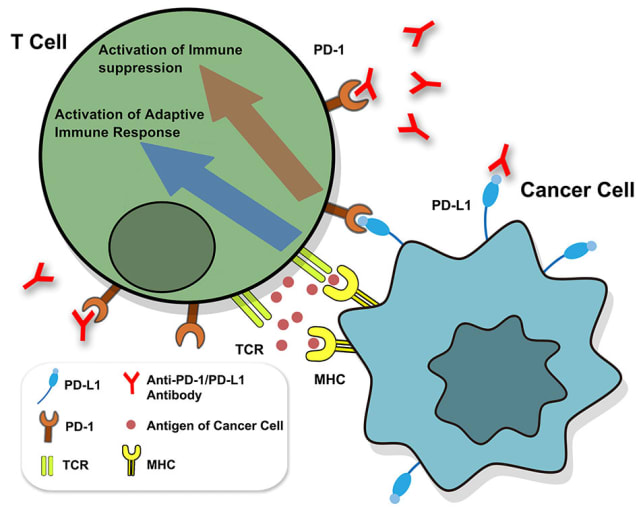
Though PD-L1 is the adversary in this story of cancer immune evasion, there is an entire troop of supporting players and a world of complex activity behind its expression. What molecular regulators—from small soluble proteins to transmembrane proteins to transcription factors—control both mRNA and surface protein levels for PD-L1? When and where do these regulators function, pre- or post-transcriptionally? Additionally, how do these molecular pathways broadly influence cancer cell phenotypes and T-cell activity?
To untangle these complex molecular circuits—operating across multiple omic levels, including the genome, proteome, and transcriptome—and identify potential novel targets for therapy, a team of scientists took a unique experimental approach. Two groups, one out of the New York Genome Center led by Dr. Rahul Satija, and another out of the Broad Institute of MIT and Harvard led by Dr. Aviv Regev, recently employed multimodal single cell CRISPR screening techniques called ECCITE-seq and Perturb-CITE-seq, respectively, to simultaneously measure transcriptomes, surface protein levels, and genetic perturbations at single cell resolution. These methods allowed them to rapidly screen the functional effects of hundreds of individual genetic perturbations—edits in the genome of a cancer cell that can knock out or interfere with a specific gene—on the expression of immune checkpoint molecules like PD-L1. While other CRISPR screening techniques would’ve necessitated hundreds of separate cell lines, each with individual edits to link perturbations with their phenotypic effects, this pooled approach allowed them to scale and screen hundreds of different gene targets across the entire cellular pathway at once to fully assess the pathway effects on ICI response.
With this rich, multiomic data, they sought to reveal cellular mechanisms of immune checkpoint resistance, as well as provide information on the downstream function of the transcriptional products of perturbed genes, including when and how they interact with PD-L1, whether pre- or post-transcriptionally. In the sections below, explore these two studies and learn how each team leveraged single cell CRISPR screens to make new discoveries.
Unraveling the circuity of IFN-γ stimulation and PD-L1 expression
Interferon gamma (IFN-γ) is a cytokine known to induce PD-L1 expression in cancer cell lines and in the tumor microenvironment (1). But with what molecular regulators does it partner to drive this activity in cancer cells? To solve this question, a team out of the Satija Lab led by graduate student Efthymia Papalexi developed a cancer cell line model to identify the genes involved in PD-L1 expression after IFN-γ stimulation. Analyzing stimulated and unstimulated cells with simultaneous single cell transcriptional profiling and detection of surface protein expression for key immune checkpoint molecules, they identified a set of 200 genes for which expression correlated with immune checkpoint surface protein induction. Of these, 26 were selected for further downstream characterization, including 18 genes that until now had no known link to PD-L1 regulation.
To functionally characterize these genes, the team designed a pooled single-guide RNA (sgRNA) library to perturb each of these genes. After initiating the gene perturbations in their cancer line, cells were stimulated with IFN-γ to engage the ICI pathway and assess how perturbations affected cellular responses. Completing their single cell CRISPR screening workflow, they were able to simultaneously detect gRNAs alongside cellular transcriptomes and surface protein expression of immune checkpoint molecules for over 22,500 single cells. This rich dataset was then used to identify putative molecular regulators affecting PD-L1 expression.
They identified a new role for the ubiquitin ligase CUL3, whose knockout perturbation increased PD-L1 protein levels. Specifically, the team noted a relationship between the CUL3 knockout and enrichment for target genes of the NRF2 signaling pathway, a known transcriptional activator of PD-L1. They hypothesized the CUL3 perturbation may disrupt the CUL3–KEAP1 complex thus interfering with the ubiquitination of NRF2, boosting pathway activation and PD-L1 transcript expression. These insights have further clinical relevance, as the study authors noted that KEAP1 is frequently mutated in lung cancer, while mutations in NRF2 and KEAP1 are associated with treatment resistance (1).
You can read more about this study here.
How CD58 enhances resistance to T-cell-mediated killing
Another team of researchers sought to survey the genes that help cancer cells evade the T-cell activity unleashed by cancer immunotherapy, this time leveraging a unique cell culture model in which malignant cells derived directly from patient tumors are cultured along with their own tumor infiltrating lymphocytes (TILs). Led by Dr. Chris J. Frangieh of the Broad Institute of MIT and Harvard, the team established this patient-derived melanoma cell culture transduced with a pooled CRISPR guide library targeting 248 putative immunotherapy-resistance genes. The cancer cells were then co-cultured with TILs, and subsequently treated with IFN-γ. Using surviving cancer cells, they next performed the Perturb-CITE-seq screen for functional evaluation of enriched perturbed genes.
They noted some unexpected patterns, including enriched perturbations for surface protein molecules like CD47, CD58, and CDH19. In particular, they noted that knocking out CD58—an adhesion molecule typically expressed on antigen-presenting cells—not only conferred resistance to T-cell killing in cell populations enriched with that perturbation, but generally, co-culture had reduced levels of CD58 RNA and protein. Additionally, knocking out CD58 was observed to drive increased expression of PD-L1. CD58 operates independently of the IFN-γ–JAK/STAT pathway, known to be disrupted in clinical models of cancer resistance, suggesting it may represent an alternative resistance mechanism that could work directly by disrupting adhesion between T cells and cancer cells presenting antigen, or indirectly by increasing the expression of a key surface protein for cancer survival, PD-L1 (2).
Find more details about how this team of researchers leveraged single cell CRISPR screening here.
Unearthing every survival mechanism to better fight cancer
Fully understanding complex molecular circuits, such as the diverse survival mechanisms employed by cancer cells, requires multimodal functional genomic screens that provide comprehensive characterization of the layers of biology, from genes to transcripts to surface proteins, involved in those circuits. As these studies demonstrate, single cell CRISPR screening will be a valuable tool to unearth all of the ways cancer cells avoid T-cell-mediated killing and what molecular regulators, whether a direct driver or an indirect backup mechanism, participate in upregulating PD-L1 expression. With these new findings and more to come, there is hope to not only unravel the drivers of varying responses to current treatments, but also to discover new therapeutic targets and treatment strategies that can make the immunotherapies we know today even more effective.
References:
- Papalexi E, et al. Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens. Nat Gen 53: 322–331, 2021.
- Frangieh CJ, et al. Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat Gen 53: 332–341, 2021.