Introducing Chromium Single Cell Multiome ATAC + Gene Expression

ATAC plus gene expression, together at last
Single cell gene expression has revolutionized how we think about biology, and cellular heterogeneity in particular. Rather than accepting an average snapshot of how cells work, scientists now have the ability to tease apart how seemingly homogenous cell lines can respond differently to the same agonist or how a defect in a rare cell type, that would otherwise be masked by its neighbors, can contribute to disease.
Despite how much positive press the transcriptome gets, however, it is ultimately the byproduct of a highly coordinated program of gene expression regulated by the epigenome. The epigenome is informed by your DNA’s chromatin state, which can be open (and accessible) or closed (and inaccessible). Chromatin state impacts how DNA-binding proteins like transcription factors or RNA polymerase can interact with genomic DNA, meaning that epigenomic regulation not only informs developmental decisions, disease progression, and therapeutic response, but also often precedes transcriptional changes and can be used to unravel otherwise indistinguishable cell types.
Researchers have leveraged Single Cell ATAC (Assay for Transposase Accessible Chromatin) to characterize regulatory elements in their samples. Now, in an effort to gain even deeper insights into cell identity, many are looking to access a unified view of a cell’s open chromatin landscape and gene expression profile. Currently, some researchers split their samples, processing half with single cell RNA-seq and half with single cell ATAC-seq, to get both transcriptome and epigenome. Then they infer which cell types correspond between the two datasets. However, this method can be heavily reliant on assumptions about the relationship between transcription start site, promoter location, and gene expression levels, as well as a priori information about epigenetic profiles of different cell types. Further, differences that arise from the potentially disparate workflows by which the two samples are processed may impact cell state and recovery. Imagine, instead, that you could assay both modalities from the same exact cell at the same time. No inference required, no waiting for a bioinformatician to see your paired results, and no fights started over which assay gets priority when the cell count is low.
The new Chromium Single Cell Multiome ATAC + Gene Expression lets you do just that. Capturing both transcriptome and epigenome simultaneously in the same single cells, this product enables the insights you need to characterize how gene expression patterns, cell types, and states are established. Samples can be analyzed with greater depth and confidence, leading to the discovery of new gene regulatory interactions or a better interpretation of epigenetic profiles with key expression markers.
For example, imagine a cancer researcher wants to understand the mechanisms behind therapeutic resistance. By profiling both mRNA expression and chromatin landscape in the same cells from responsive and non-responsive patients, they could identify tumor-specific activation of transcription factors and key regulatory elements, then link them to their target downstream genes through the associated changes in gene expression. These insights could help to resolve cancer cell–specific gene regulatory programs and gene expression signatures that give rise to resistance. But this is just a single use case. How could Single Cell Multiome ATAC + Gene Expression help uncover the gene regulatory networks driving immune cell lineage differentiation, or identify target genes and cell types associated with neurodegeneration and psychiatric disorders? What deeper insights could it enable for you?
So, how does it work?
To understand how this dual-modality processing is possible, we spoke with two 10x Genomics R&D scientists involved in the development of Single Cell Multiome ATAC + Gene Expression: Kamila Belhocine, Staff Scientist; and Shamoni Maheshwari, Senior Computational Biologist.
We learned that Chromium Single Cell Multiome ATAC + Gene Expression begins with a single nuclei suspension. Transposition is performed in bulk using the enzyme transposase, which preferentially cuts nuclear DNA in open chromatin regions. Transposed nuclei are then partitioned into droplets, or GEMs, with a single Gel Bead that contains a unique 10x Barcode. Within the GEM, the unique barcodes are attached to available mRNA and transposed DNA fragments in a single nucleus. Following this incubation, GEMs are broken and pooled before clean up, pre-amplification, and library construction. Two libraries are made from a single pool of GEMs, one for sequencing RNA and one for ATAC.
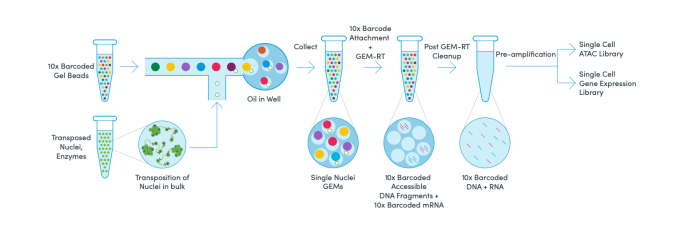
After learning more about the workflow, and how this technology is enabled, we asked how they see multiomics being used to resolve biological questions.
What research problems were you trying to solve in developing Chromium Single Cell Multiome ATAC + Gene Expression?
Kamila: For the past few years, measuring gene expression at the single cell level has empowered scientists to study and understand their biological systems like never before. Many of these gene expression patterns are regulated by the epigenetic programs coded by changes in chromatin accessibility, which can be measured by ATAC-seq. However, while differences in chromatin accessibility between cell states are prominent and readily detected, they can be challenging to interpret without a direct readout of the effects of these epigenetic changes on gene expression. Single Cell Multiome ATAC + Gene Expression is the first commercial solution that can elucidate the precise interactions between chromatin accessibility and gene expression to enable an understanding of gene regulation in health and disease at the highest resolution.
Can you describe a powerful feature or capability of the software analysis pipeline, Cell Ranger ARC?
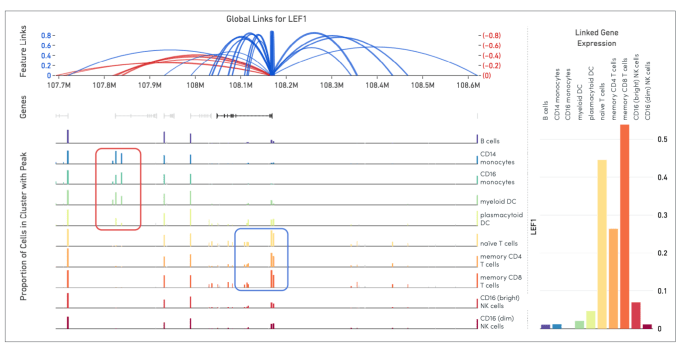
Shamoni: The Human Genome Project determined that 1% of the human genome is composed of genes. In the following decade, the Encyclopedia of DNA Elements (ENCODE) project identified nearly 20% of the human genome as having potential regulatory function. Cell Ranger ARC software has the power to bridge these potential regulatory regions to the transcriptome by identifying correlations between open chromatin sites and gene expression in single cells.
What discoveries do you think scientists can now make that weren’t possible before?
Shamoni: Bulk assays such as the ones carried out by ENCODE have generated a catalog of regulatory elements across the human genome. By applying this method on a variety of different samples and cell types, researchers will be able to parse precisely which cell types/states a regulatory element is active in and the expression status of its target gene.
Cells are usually divided into static buckets called “cell types” by drawing arbitrary boundaries based on differences in the observed gene expression profiles. However, we know that, in vivo, cells occupy a continuum of states and gene expression is the terminal readout controlled by a variety of epigenetic factors. This analysis adds information on putative regulatory status of the gene expression program in a cell, thereby enabling classification into dynamic cell states.
Finally, scientists can improve interpretation of non-coding variation. The majority of SNPs identified in GWAS studies fall in non-coding regions. The challenge then is to interpret this genetic variation and identify its functional target. This assay has the power to reveal, if a non-coding SNP falls under an open chromatin peak, whether that putative regulatory region is active in the specific cell type of interest and what genes it is potentially regulating.
Kamila: With Single Cell Multiome ATAC + Gene Expression, scientists will be able to establish the mechanisms of gene regulation in health and disease, better understand the effects of genetic disruptions in non-coding regions, and uniquely contribute to the extensive knowledge base that will ultimately result in new therapies improving people’s lives.
How does it feel when you see scientists making discoveries with a product that you helped to develop?
Kamila: As product developers, we strive to understand our users’ needs and build products to robustly address them. I am always amazed by the discoveries enabled by our products and their impact on our collective understanding of disease and development. I truly believe that a better understanding of biology will ultimately result in improving the health and quality of life of millions to billions of people, and with each new publication that uses products we developed, I feel that 10x Genomics contributes in a meaningful way towards that goal.
How would you use it?
While scientists at 10x Genomics love to imagine ways our products could be used to advance human health, what we enjoy even more is learning what you are already doing (or going to do) with them. With that in mind, we’d love to hear from you. What questions do you plan to answer with Single Cell Multiome ATAC + Gene Expression? Are there new research areas that multiomics will open up for you? Follow us on Twitter to see what other scientists are saying.
To find out more about Single Cell Multiome ATAC + Gene Expression, including product videos, resources on the workflow, and datasets for download, visit the product page.
And check out our recent webinar, introducing Single Cell Multiome ATAC + Gene Expression and its capabilities. Watch on-demand.