Introducing the Innovator Series: Hacking ATAC-seq to perform clonal lineage tracing in tumors

What is the Innovator Series?
Every once in a while, a new technology, an old problem, and a big idea turn into an innovation.
- Dean Kamen, American engineer, inventor, and businessman
Scientists are searchers and problem-solvers. When they face barriers to their investigations, they roll up their sleeves to find the solution, turning challenges into opportunities to drive the leading edge of what’s possible. The Innovator Blog Series celebrates research conducted by 10x Genomics customers who have demonstrated scientific ingenuity by adapting Chromium Single Cell or Visium Spatial sequencing-based assays. These innovations are distinguished by their originality and potential impact on scientific discovery, including providing access to novel analytes, advancing multiomic analysis techniques, and demonstrating critical applications to human health and disease research. These techniques are customer developed, meaning they are not officially supported by 10x Genomics.
In our first Innovator Series article, we profile research conducted by Dr. Caleb Lareau, Postdoctoral Research Fellow in the Satpathy Lab at Stanford University. At our recent Global Virtual Cancer Symposium, he shared a new technique called mitochondrial single cell ATAC-seq (mtscATAC-seq), which adapts the 10x Genomics single cell assay for transposase-accessible chromatin sequencing (scATAC-seq) to capture mitochondrial DNA from tumor cells. Read on to learn how this method helped Dr. Lareau and his colleagues assess tumor clonal heterogeneity—a major driver of tumor progression, evolution, and metastasis—through clonal lineage tracing.
Understanding tumorigenesis through mitochondrial DNA
“The basic innovation is the demonstration that neoplasia is discontinuous in space and time; it is a dynamic process advancing through stages that are qualitatively different” (1).
In 1958, Dr. Leslie Foulds of the Chester Beatty Research Institute, Institute of Cancer Research, Royal Cancer Hospital, wrote these words, establishing a new model of tumorigenesis in which cancer does not, as previously speculated, progress in a regular, linear fashion. Rather, as Foulds determined, cancer arises from a more complex, diverging path (2). Researchers in the 1970s would go on to propose an evolutionary model of cancer progression driven by genomic instability, resulting in heterogeneous cancer cells branching away from a clone of origin (2).
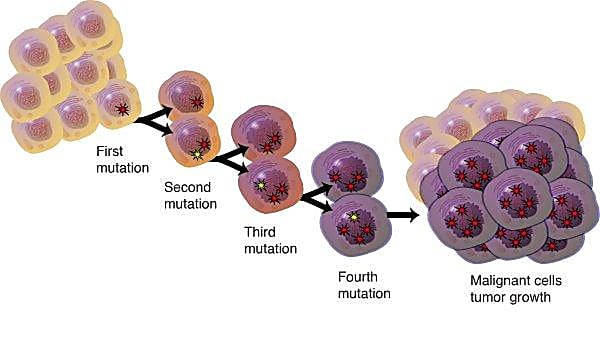
This heterogeneity is now thought to play an essential role in tumor progression, variable responses to therapy, and relapse. According to Caswell and Swanton, “insight into the clonal complexity of individual tumours and its contribution to progression, immune evasion and exhaustion may be critical to the development of more effective cancer therapeutics” (2).
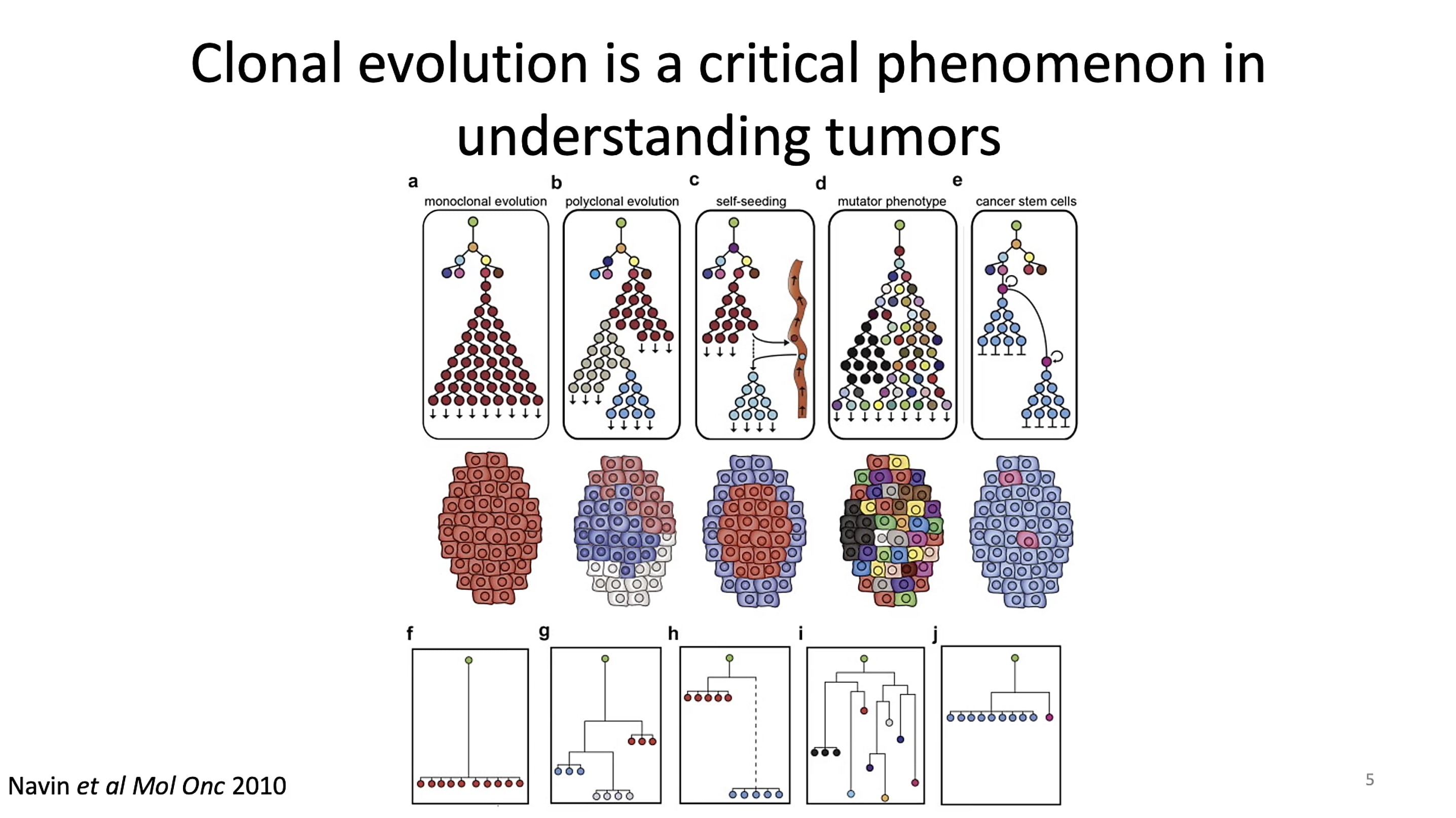
But understanding the cell-to-cell changes that drive tumor progression can be challenging when the hallmarks of change are difficult to identify. Researchers have leveraged genetic mutations to follow the path from clone of origin to tumor, including copy number variants (CNVs) and single nucleotide variants (SNVs). In his talk at the Global Virtual Cancer Symposium, Dr. Lareau noted, however, this approach breaks down when working with tumors that don’t have a wide amount of detectable CNVs or SNVs. This challenge led his team to consider alternative approaches to deconstruct clonal lineage and uncover mixed cell populations in tumors.
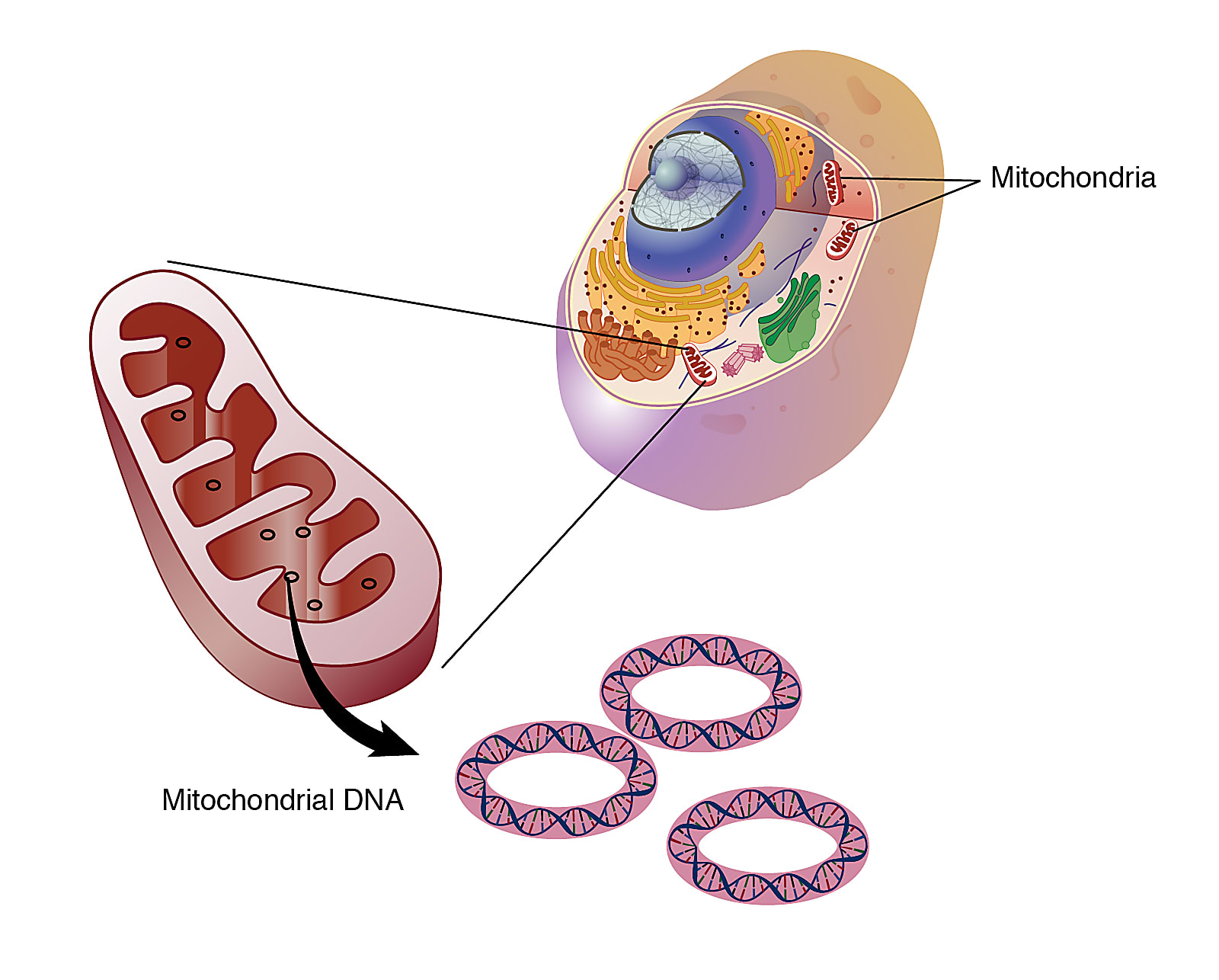
Dr. Lareau landed on somatic mitochondrial DNA (mtDNA) as an effective biological parameter to trace the clonal lineage of tumor cells. mtDNA is passed down to daughter cells at each cycle of cell division, and is heavily mutagenized, at 10 to 100 times the mutation rate seen in the nuclear genome. This makes mtDNA highly valuable to trace somatic mutations in the cell lineage. Experimentally, as well, mtDNA is very accessible. There are hundreds to thousands of mtDNA copies in each cell, allowing high confidence to define mutations. And because each molecule is 200,000 times smaller than the nuclear genome, sequencing mtDNA is not cost-prohibitive.
Dr. Lareau noted another key benefit of using mtDNA: it could be measured through a standard chromatin accessibility protocol.
“In the original ATAC-seq chemistry, as much as half of the reads will be attributed to mitochondrial DNA. This is because...mitochondrial DNA does not have any nucleosomes or histones, so the entire 16.6 kilobase contig is essentially sitting there in the cytoplasm, in the mitochondria, accessible for Tn5 [transposase] to cut. [...] This was thought of as a nuisance as part of the ATAC-seq chemistry, but for us interested in sequencing mitochondrial DNA in order to create depth, really this provided the best of both worlds in that using the standard ATAC-seq chemistry gives a very deep sequencing of mitochondrial DNA, which we’re going to evaluate for somatic mutations as well as accessible chromatin for inferring properties of cell state.”
With the ability to perform deep mitochondrial sequencing, Dr. Lareau explained that they could detect even low frequency somatic mutations, possibly revealing hidden drivers of clonal evolution or small populations of divergent tumor cells that contribute to heterogeneity.
A sample prep hack to leverage single cell ATAC-seq
Because every cell counts when it comes to tumor heterogeneity, a single cell view of mitochondrial DNA was essential for Dr. Lareau’s experiments. This led him and his colleagues to the 10x Genomics Chromium Single Cell ATAC-seq assay, albeit with one small catch: ATAC-seq requires single nuclei as the sample input, which would mean losing the mitochondria.
Dr. Lareau’s next challenge was to develop a protocol that could retain the cytoplasm and its contents in a droplet-based single cell ATAC-seq reaction. Three key innovations to the sample prep and data processing made this possible. In particular, they implemented a modified cellular lysis protocol to retain mtDNA, added an extra fixation step, and developed a new computational framework to call variants, including variants lower in frequency. These innovations allowed Dr. Lareau and his colleagues to achieve nearly unbiased coverage across the entire mitochondrial chromosome at single cell resolution, with overall higher cell throughput and performance.
The advantages of this approach were also clear when applied to a chronic lymphocytic leukemia sample. Individuals with leukemia have a high count of B cells in the peripheral blood, which typically share one predominant B-cell receptor (BCR) sequence. This is a sign of a clonal expansion, meaning a majority of the B cells that compose the tumor are the daughter cells of a clone of origin which sets the receptor sequence. Dr. Lareau and his colleagues wanted to see, however, if they could find populations of unique subclones among these seemingly homogeneous B cells.
For half of their leukemia samples, they used 5’ single cell RNA-sequencing technology from 10x Genomics to measure gene expression and BCR sequences of individual cells. For the remaining half of their samples, they leveraged the adapted mtscATAC-seq protocol to identify subclonal mitochondrial DNA mutations. One patient, in particular, had a dozen somatic mutations, allowing them to cluster the B cells into 9 unique subclones.
“After finding these mutations, we sort of wondered, is there any functional consequence of these tumor cells tagged by these mutations?”
Taking a closer look at a group of cells for which they sequenced both the BCR sequences and mtDNA mutations, they refined potential subclone populations. While some subclones carrying mitochondrial mutations had the same BCR sequence as the majority of the tumor, one small population of B cells shared a mutation at the 14858G allele and had a different BCR sequence.
“Ultimately, this provides some overall corroboration that the DNA mutation is marking some sort of real, functional subclone that has a very distinct BCR sequence from the rest of the tumor.”
Unlocking greater multiomic potential with mtscATAC-seq
Providing a high-resolution view of the underlying biology that defines unique subclones in a tumor, mtscATAC-seq has the potential to help scientists build a more comprehensive knowledge of cancer by unlocking hidden tumor heterogeneity. And this is just the beginning. Continuous innovation in next-generation multiomics—the capture of multiple measurements simultaneously from the same single cell or tissue section—is opening up new doors, adding on more analytes to the ones scientists are already able to access in the same experiments. Dr. Lareau and other leaders in the field, Drs. Eleni Mimitou and Peter Smibert, formerly of the New York Genome Center, worked together to develop ASAP-seq (ATAC with Select Antigen Profiling by Sequencing). Harnessing the methodological advances demonstrated by mtscATAC-seq, namely the additional cell fixation step to preserve protein expression, ASAP-seq allows scientists to “collect information about chromatin accessibility, abundance of hundreds of cell surface and intracellular proteins, and mtDNA variants for thousands of single cells” (3). Soon after, the team also developed DOGMA-seq, which leverages Chromium Single Cell Multiome ATAC + Gene Expression from 10x Genomics to capture one more layer of information, RNA, from the same cell.
You can learn more about these innovations here →
And, to watch Dr. Lareau’s full presentation on mtscATAC-seq on-demand, visit our Global Virtual Cancer Symposium hub.
It’s incredible and humbling to know that 10x Genomics customers are driving the future of single cell multiomics. Until the next Innovator Series blog, we look forward to seeing the experimental challenges you overcome and the new depths of insights you reach.
References:
- Foulds L. The natural history of cancer. J Chronic Dis 8: 2–37 (1958).
- Caswell DR and Swanton C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med 15: 133 (2017).
- Tang L, et al. Arsenal of single-cell multi-omics methods expanded. Nat Methods 18: 858 (2021).