Resolving the transcriptional events of life’s earliest stages with CRISPR and single cell RNA-sequencing

Co-authored by Dr. Celia Alda-Catalinas, Genome Editing Scientist at GSK, Stevenage, UK
Walk through Dr. Celia Alda-Catalinas’ research process as she combined single cell RNA-sequencing with functional CRISPR screens to identify 44 epigenetic regulators of a key transcriptional activation event during mammalian embryonic development. This work was conducted at the Epigenetics Programme of the Babraham Institute, Cambridge, UK, led by Dr. Celia Alda-Catalinas and supervised by Dr. Melanie Eckersley-Maslin, Dr. Oliver Stegle and Professor Wolf Reik.
Mammalian development is initiated upon formation of the zygote. The egg is fertilized, setting in motion a number of cell division and differentiation events that lead to formation of the implanting embryo. Underlying these events are a series of regulated changes in gene expression. One major transcriptional event is called zygotic genome activation (ZGA), and was a process Dr. Celia Alda-Catalinas, formerly a member of the Epigenetics Programme at the Babraham Institute in Cambridge, UK, sought to understand while conducting research for her doctoral degree.

ZGA is a critical transcriptional activation event in mammalian embryonic development, marking the moment when the zygote’s own genes become expressed. Though the genes that are turned on during ZGA are relatively well characterized, the upstream factors that regulate their expression are not. As Alda-Catalinas explained, “mammalian genomes undergo drastic changes in their epigenome after fertilization, to reprogram from fully specialized germ cells to a zygote that needs to be able to develop into a full organism.”
These uncharacterized changes in the epigenetic landscape motivated her research efforts to find maternally inherited proteins accumulated in the embryonic cytoplasm that could serve as epigenetic regulators of ZGA. These proteins would subsequently enter the nucleus to interact with genomic DNA and manipulate transcription after fertilization. “Because ZGA is a transcriptional activation event where gene expression goes from 0 (meaning the zygotic genome is initially quiescent) to expression of thousands of genes, we were looking for regulators that are activators,” said Alda-Catalinas. From a list of known proteins expressed in oocytes and zygotes, she and her colleagues selected 230 candidate genes to perform further functional testing on an in vitro model. Knowing that a small population of mouse embryonic stem cells (mESCs) resembles the two-cell mammalian embryo in both the transcriptome and epigenome at the time of ZGA, they used mESCs to study if overexpression of the selected maternal candidates could induce a ZGA-like gene expression signature in the mouse cells. “Consequently, we needed a system to upregulate our candidate genes rather than repress them.”
How to perform CRISPR-activation screens to identify epigenetic regulators
Alda-Catalinas and her colleagues found the system they needed in CRISPR-Cas9. Though this technology is notorious for its ability to create targeted cuts in the genome, catalytically inactivated CRISPR-Cas9, or dCas9, can also be used to repress or activate transcription in genes of interest. These methods are called CRISPRi and CRISPRa, respectively (1).
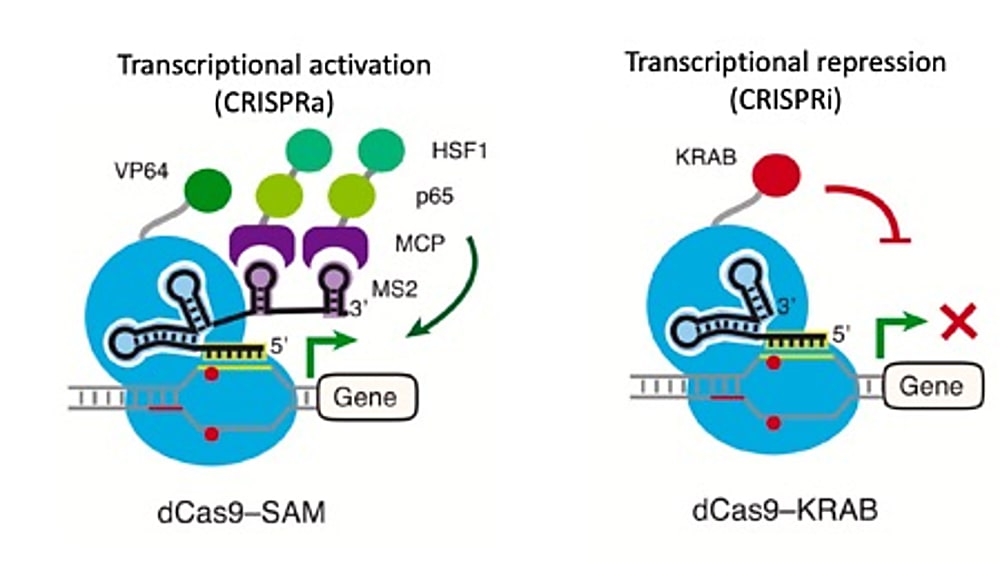
The system utilizes short single guide RNA sequences, or sgRNAs, to direct dCas9 proteins fused with effector domains to specific endogenous genes, where transcription is subsequently repressed or activated depending on the effector domain function (1). Critically, the ability to control the expression of individual transcripts through a targeted approach enables exploration of not only a broader range of protein-coding genes, but also less understood, non-canonical transcripts (1), including upstream factors like those Alda-Catalinas and her colleagues were interested in.
They first engineered a stable CRISPRa mESC line. This entailed introducing a dCas9-activator fusion protein and an RNA-binding activator fusion, dCas9-VP64 and MS2-p65-HSF1, respectively, into the stem cells through lentiviral transduction, a method of viral delivery. Through clonal selection, they generated a clonal cell population that would go on to host the genomic events of their CRISPR-activation experiment.
Alda-Catalinas explained that their sgRNAs were designed to include specific features, including two MS2 loops in the scaffold sequence of the sgRNA, that would help recruit the p65-HSF1 trans-activators via fusion to the MS2 RNA binding protein.Planning for 2 sgRNAs per candidate gene and 15 non-targeting control sgRNAs, making a total of 475 sgRNAs covering 230 candidate genes, Alda-Catalinas and her colleagues prepared a standard pooled sgRNA library. The oligo sgRNA library was PCR-amplified, then cloned into the modified sgRNA-MS2 lentiviral backbone using a cloning procedure called Gibson assembly, and finally packaged into lentivirus. This library was then transduced into the stable CRISPRa mESC line, once again through lentiviral transduction.
Following these steps to perform a CRISPR-activation screen, the team analyzed the gene expression profiles of each cell using single cell RNA-sequencing. Alda-Catalinas explained, “by reading the gene expression profiles of each perturbed cell using the 10x Genomics solution for single cell gene expression, we reasoned that we could identify epigenetic factors amongst our candidates whose over-expression by CRISPRa triggered a gene signature that resembled that seen in a two-cell embryo undergoing ZGA.” To do this analysis, Alda-Catalinas recruited the help of two computational biologists on her team, Danila Bedikhin and Oliver Stegle, who performed multiomics factor analysis (MOFA) on approximately 200,000 sequenced single cells to identify relevant gene signatures to the ZGA state in an unbiased manner. “Indeed, we did identify 44 transcription factors and chromatin remodelers whose CRISPR-activation induced a ZGA-like signature in mESCs” (3).
Benefits of combining CRISPRa with single cell RNA-sequencing
Alda-Catalinas started this project in 2016, when she was a graduate student in the Reik Lab at the Babraham Institute. At the time, “CRISPR screens were only just starting to emerge as a powerful tool for functional genomic studies,” she said. “However, we soon realized that this approach could provide a comprehensive, systematic, and novel view into the regulation of ZGA.” In this case, CRISPRa screens allowed her to understand how the quantitative expression of certain genes affected cellular phenotype. And in turn, she was able to identify what genes may serve as transcriptional regulators. While these same kinds of screens can be performed with cDNA libraries, Alda-Catalinas pointed out a few additional benefits of the CRISPR approach: “Since CRISPRa triggers transcriptional activation of the endogenous locus, the expression changes induced to the target gene and downstream are possibly more physiologically relevant than the acute overexpression induced with cDNA transfections. Moreover, CRISPRa does not require cloning of the cDNAs of interest, making it amenable for transcriptional upregulation of any gene, regardless of its size.”
As Alda-Catalinas explained, the ability to combine CRISPRa with single cell RNA-sequencing, a technique she adapted from a method called CROP-seq (4), allowed her to detect each CRISPR perturbation and its linked, quantitative gene expression read-out for her candidate genes at greater scale, throughput, and ease than other methods. “We needed to implement a complex read-out that allowed us to analyze the expression of thousands of genes to have a comprehensive understanding of a complex transcriptional event, as ZGA is. Therefore, we needed an RNA-sequencing read-out.” With this read-out at the resolution of single cells, Alda-Catalinas gained “substantial advantages in terms of scalability and the possibility to disentangle cell-to-cell heterogeneity,” compared to bulk methods.
Continuing, she explained, “we could have done individual, arrayed perturbations for the 230 candidates followed by bulk RNA-sequencing for each of them. However, considering we used 2 sgRNAs per candidate gene and 15 non-targeting controls, this would have involved the production of 475 individual lentiviruses, 475 individual transductions and cell cultures, and preparation and sequencing of 475 individual bulk RNA-sequencing libraries. Using pooled CRISPR screening, we could test the 475 perturbations in a single batch, increasing the throughput of the experiment and reducing the hands-on time. We found that the adaptation of CROP-seq to detect both the guide RNA and the whole coding transcriptome in each individual cell was the most effective and comprehensive way to interrogate the 230 candidates in a pooled CRISPRa screen.”
Tips to perform a successful CRISPR-activation screen
Science is complex, and the methods we use to study it are too. However, this complexity can be tackled and managed with a dedication to discovery and rigorous experimental planning and practice. Alda-Catalinas optimized various technical aspects of the CRISPR-activation technique before performing her screen in mouse stem cells. She offered some expert tips to aspiring scientists, interested in exploring this technique for their own research projects.
Think first. Alda-Catalinas recommended that the researcher zoom out and think about what methods and technology platform will best suit their questions of interest. She said, “First, think which is the relevant CRISPR platform to use (such as CRISPR KO, CRISPRi, or CRISPRa) for your biological system and research question. Next, think about your read-out and how much information you need to make your screen meaningful and obtain relevant hits. Do you need a complex read-out such as single cell RNA-sequencing, or can you use a simpler one such as FACS-sorting based on relevant markers?” She also emphasized thinking through details about how many technical replicates to conduct and the number of cells necessary for the study.
Perform pilot tests. Critically, Alda-Catalinas performed a pilot testto determine her overall screen design (3). Explaining this choice, she noted, “we needed to test our hypothesis that CRISPRa of relevant regulators would induce a ZGA-like signature. Secondly, we needed to make sure this signature could be detected using the 10x Genomics single cell gene expression solution.” With her colleagues, she performed individual CRISPRa experiments on two well-known markers of ZGA, and ultimately validated how many cells (in this case, 400) she would have to sequence per sgRNA to confidently detect a ZGA-like transcriptional response in a potential hit.
Prepare your samples carefully. In addition to a pilot test, Alda-Catalinas noted that “careful sample preparation was crucial for good quality of the resulting data.” She optimized her methods over the course of the experiments, and in the final dataset recovered up to 14,000 good quality single cells per lane after loading 20,000 cells.
Make time for data analysis. Finally, Alda-Catalinas recommended that researchers should invest time in their data analysis. “Think of the best quality controls for your data and the statistical parameters that allow you to identify relevant hits. Once you have your list of hits, take time for validations since pooled screens can introduce false negatives and false positives.”
Dr. Celia Alda-Catalinas’ work offers a groundbreaking experimental model for functional genomics studies. Using the combined power of CRISPR and single cell gene expression profiling, she was able to discover novel insights into the identity and function of key epigenetic regulators in a complex transcriptional event during mammalian embryonic development. Beyond this application, Alda-Catalinas sees this combined method revealing insights into disease as well. She has recently moved to the Functional Genomics team at GlaxoSmithKline (GSK) where she performs functional CRISPR screens to identify relevant disease targets. We look forward to what she and her colleagues will discover next.
On behalf of 10x Genomics, we would like to thank Dr. Alda-Catalinas for her exciting and useful contributions to this article and her continued efforts to advance the application of single cell technology for functional genomics.
For more information about how to combine CRISPR and single cell gene expression methods, take a look at our Feature Barcode Technology for CRISPR screens.
References:
- LA Gilbert et al., Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 159, 647–661. (2014).
- A Lo, L Qi, Genetic and epigenetic control of gene expression by CRISPR-Cas systems. F1000Res. 6, F1000 Faculty Rev-747. (2017).
- C Alda-Catalinas et al., A single-cell transcriptomics CRISPR-activation screen identifies new epigenetic regulators of zygotic genome activation. bioRx. (2019).
- P Datlinger et al., Pooled CRISPR screening with single-cell transcriptome readout. NatMethods. 14, 297–301. (2017).