Insights into immuno-oncology enabled by single cell sequencing

Guest author: jamie-schwendinger
Recent studies demonstrate the efficacy of single cell sequencing in the search for immune checkpoint therapy targets and treatments.
Immuno-oncology has transformed how we can treat cancer and extended hope to many with previously incurable cancers. In recognition of advances in the field of immuno-oncology, two pioneers were awarded the 2018 Nobel Prize in Physiology or Medicine “for their discovery of cancer therapy by inhibition of negative immune regulation” (1). Independently, Drs. James P. Allison and Tasuku Honjo discovered that inhibition of key immune checkpoint regulators could release the brakes on patients’ own immune systems, enabling an effective immune response to cancer and extending life. This type of immunotherapy constitutes a paradigm shift in cancer treatment, where physicians can now directly activate a patient’s immune system to fight their own cancer.
Identifying immune checkpoint regulators
CTLA-4 and PD-1 are the first immune checkpoint regulators targeted for cancer immunotherapy. They are both receptors expressed by T cells after T cell-receptor (TCR) engagement and act to dampen T cell activation and immune responses. Immune checkpoint therapy works by blocking these or other immune checkpoint regulators.
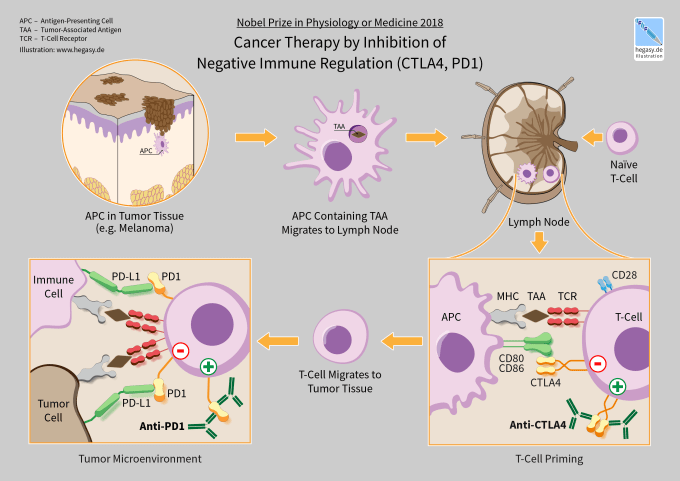
Despite the progress enabled by immune checkpoint therapy, particularly with combination therapy targeting both CTLA-4 and PD-1, numerous difficulties remain. Only a subset of cancers respond to immune checkpoint therapy, while many others, like glioblastoma, remain resistant. Activation of the immune system also comes with its own range of side effects that can impact up to 80% of patients and range from mild skin irritation to life-threatening colitis (2). Understanding how immune checkpoint therapy works, and why and when it will be effective, is crucial to deciding treatment options, and developing novel therapies moving forward.
Blocking CD73 and PD-1 prove a potent combo
To investigate why glioblastoma resists immune checkpoint therapy, Padmanee Sharma’s lab at the University of Texas MD Anderson Center used mass cytometry (CyTOF) to identify the immune populations present in different tumor types, comparing cancers responsive to immune checkpoint therapy, such as non-small-cell lung carcinoma, to glioblastoma multiforme (3). They found that glioblastoma tumors had a unique population of CD68+ myeloid cells that were also high expressors of CD73 (CD73hi), along with classic immune checkpoint inhibitors like PD-1. Though CD73 had previously been shown to contribute to immunosuppression in glioblastoma, the role of these CD73hi cells was unknown.
To better understand the role of this population of cells, Professor Sharma and her collaborators then used the Chromium Single Cell Gene Expression Solution to characterize the transcriptome of all cells within glioblastoma tumors. Armed with a key gene expression signature describing the unique CD73hi subset of the CD68+ myeloid population, they found that the presence of these cells correlated with lower overall survival and CD73hi clusters persisted after treatment with anti-PD-1. Further, they found that the CD73hi cluster expressed several receptors that are currently targets in clinical trials for treatment of gliobastoma multiforme, making CD73 potentially a more relevant therapeutic target, since the majority of cells expressing the putative targets also expressed high levels of CD73 (3).
The importance of CD73 as an immunotherapy target was confirmed using a mouse model of glioblastoma. Mice lacking CD73 showed reduced tumor growth compared to wild type. Excitingly, the loss of CD73 coupled with combination immune checkpoint therapy using anti-CTLA-4 and anti-PD-1 resulted in significant survival benefits compared to untreated CD73-/- or immune checkpoint therapy alone. Given the narrow specificity of cases in which immune checkpoint therapy is effective, identification of improved combination therapy could have major clinical implications.
Expanding the realm of immune checkpoint regulators with multiomic analysis
VISTA is a unique immune checkpoint regulator, and VISTA blockade has been shown to reduce tumor growth (4). While CTLA-4 and PD-1 are expressed during inflammation on activated T cells, VISTA is constitutively expressed on naive T cells and lost under inflammatory conditions. To better understand the role of VISTA in immune tolerance (rather than cancer therapy), Mohamed A. EITanbouly, Yanding Zhao, and colleagues at Dartmouth specifically deleted VISTA expression in CD4+ T cells and then looked at transcriptomic and epigenomic changes that resulted from VISTA loss. To do this, they used both the Chromium Single Cell Immune Profiling Solution and Chromium Single Cell ATAC Solution. The assay for transposase-accessible chromatin (ATAC) gives a genome-wide picture of epigenomic changes within the cell by identifying cut sites where a modified transposase enzyme cleaves open DNA. In regions of silent, or closed, chromatin, and where promoter elements or enhancer proteins are bound, the transposase cannot access the DNA, and no cleaved fragments are produced. ATAC, therefore, can provide a picture of a cell’s current state, as well as what state a cell is poised to enter, by giving insight into what promoters are bound and what genes are available to be transcribed—even if they haven’t been transcribed yet.
By combining information about gene expression levels, TCR sequence, and chromatin state, the researchers found that mice lacking VISTA in CD4+ T cells showed an alteration in proportions of naive T cells, with an expansion of memory-like T cells at the expense of quiescent cells. Further, ATAC data indicated that the memory-like T cell cluster had increased accessibility for TCR effector genes, suggesting these cells were primed to be particularly TCR-responsive. However, TCR sequencing of autoreactive T cells showed that loss of VISTA impacted neither their distribution nor percentage. Together, this demonstrated that VISTA has a unique role as the first negative checkpoint regulator specific to naive cell fate and function (5).
Single cell analysis helps explain why treatment works
CDK7 is a cell cycle regulator. Using a recently identified specific inhibitor of CDK7, Hua Zhang and co-authors at NYU Langone Medical Center and Dana-Farber Cancer Institute demonstrated that inhibition of CDK7 in mice could slow tumor progression and increase survival in accelerated disease models of small cell lung cancer (6). They next looked at treatment efficacy for combination therapy, and found that mice treated with both CDK7 inhibitor and anti-PD-1 showed a two-fold increase in survival relative to controls. While this dramatic response is encouraging, of even greater value may be understanding how this combo therapy works, and how to identify other successful combination therapies.
In order to understand the mechanism behind tumor response to CDK7 inhibition, these scientists relied on the Chromium Single Cell Gene Expression Solution alongside immune profiling with flow cytometry. They found that, as expected, inhibition of CDK7 led to a stall in cell cycle and reduction in growth, but also triggered an immune response, leading to tumor-intrinsic activation of CD8+ T cells and increases in pro-inflammatory cytokines and chemokines. Cluster analysis from single cell RNA-sequencing delineated T cells into 11 distinct groups, and demonstrated that more than an increase simply in number of CD4+ or CD8+ T cells, CDK7 inhibition and immune checkpoint therapy impact cell function so that both CD4+ and CD8+ T cells become more proliferative and adopt an activated/effector T cell signature. With this finding, the authors were able to connect the cell cycle effects of CDK7 to immune-response signaling, demonstrating the breadth and depth of information available from single cell measurements.
The potential therapeutic use of negative checkpoint regulators is just getting started. In addition to blocking CTLA-4, PD-1, VISTA, or other components of immune checkpoint regulation with antibodies or small molecules, CRISPR-Cas9 editing may be used to target these receptors. In fact, the first phase 1 human clinical trial testing the safety of multiplex CRISPR-Cas9 genome editing for cancer immunotherapy included deletion of PD-1 in transferred T cells, in order to reduce T cell exhaustion (7). The rapid movement of basic and translational research to the clinic emphasizes the potential therapeutic benefits of studies that manipulate and strive to understand the mechanisms of negative checkpoint regulators, particularly from a whole-genome, single cell perspective.
References
- https://www.nobelprize.org/prizes/medicine/2018/summary
- JM Fritz, MJ Lenardo, Development of Immune Checkpoint Therapy for Cancer. JEM. 216, 1244–1254 (2019).
- S Goswami, T Walle, AE Cornish et al., Immune Profiling of Human Tumors Identifies CD73 as a Combinatorial Target in Glioblastoma. Nature Medicine. 26, (2019).
- SL Topalian, CG Drake, DM Pardoll, Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell. 27, 450-461 (2015).
- MA EITanbouly, Y Zhao, E Nowak et al., VISTA is a Checkpoint Regulator for Naive T Cell Quiescence and Peripheral Tolerance. Science. 367, 264 (2020).
- H Zhang, CL Christensen, R Dries et al., CDK7 Inhibition Potentiates Genome Instability Triggering Anti-tumor Immunity in Small Cell Lung Cancer. Cancer Cell. 37, 1-18 (2020).
- EA Stadtmauer et al., CRISPR-Engineered T Cells in Patients with Refractory Cancer. Science. DOI: 10.1126/science.aba7365