Modern drug discovery: Single cell systems pharmacology reveals a novel combination therapy against leukemia

Childhood leukemia is the stage of a breakthrough era of drug discovery and development as scientists employ computational tools to derive deeper insights regarding drug response and resistance, mechanisms of action, and more from single cell and bulk RNA-seq data, and inform decisions on new therapeutic approaches.
In a powerful example, recently published in Cancer Cell, a global team of cancer researchers leveraged network-based analyses of single cell and bulk RNA-sequencing data from healthy human B cells and leukemia patient samples to identify development states driving resistance and sensitivity to a common chemotherapeutic agent, asparaginase, against B-cell acute lymphoblastic leukemia (B-ALL). Interrogating driver genes from the resistant subpopulation revealed a druggable protein target, BCL2, that provided a robust logic to explore combination therapy with venetoclax to overcome resistance and enhance anti-tumor effects.
Keep reading to explore the team’s unique applications of computation and single cell technologies, including Chromium Single Cell Multiome ATAC + Gene Expression and Single Cell Gene Expression Flex, to enable these potential advancements for B-ALL treatment.
Jump ahead to the following sections:
- Human Cell Atlas data maps the B-cell landscape
- Deconvoluting bulk RNA-seq leukemia patient data with single cell B-cell profiles
- Validating resistant and sensitive B-cell states with Multiome
- Identifying BCL2 as a viable target for combination therapy
- Evaluating combination therapy efficacy with Flex

Applying Human Cell Atlas bone marrow datasets to understand the B-cell developmental landscape
B-cell acute lymphoblastic leukemia (B-ALL) is a common childhood cancer that arises from excessive production of B cells in the blood and bone marrow (1). Faulty development is a potential cause of this disease, and other studies of similar cancers, including T-cell acute lymphoblastic leukemia, have suggested that the developmental stage at which blood cells turn cancerous plays an important role in influencing therapeutic responses (1).
So what stages of B-cell development give rise to B-ALL? And how do the unique properties of those cell states affect the mechanism of action of common drugs against B-ALL—or possibly influence resistance?
Seeking to answer these questions, a team of researchers from St. Jude Children’s Research Hospital and the Hefei Comprehensive National Science Center in China took a first principles approach, revisiting the healthy B-cell developmental landscape. They turned to a single cell dataset of healthy human bone marrow generated by the Human Cell Atlas. The dataset included 283,893 bone marrow cells, 54,574 of which were from the B-cell lineage, from eight donors. Clustering by marker gene expression revealed eight B-cell development states: hematopoietic stem cells (HSC), CLP (common lymphoid progenitor), pre-pro-B, progenitor B (pro-B), precursor B (pre-B), immature-B, mature-B, and plasma cells (2).
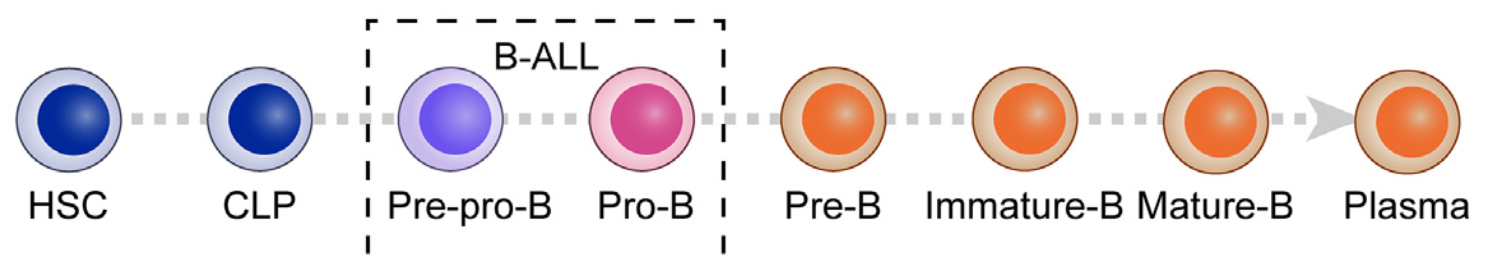
But cell-type information was not all the team obtained from this single cell dataset. Using systems biology algorithms SJARACNe (an updated version of ARACNe, Algorithm for the Reconstruction of Accurate Cellular Networks) and NetBID2 (data-driven Network-based Bayesian Inference of Drivers), the team inferred molecular interactions between the transcription factors and signaling proteins that drive development for each of these B-cell states. This analysis allowed them to identify the protein activity corresponding to known marker genes in B-cell development from single cell gene expression data, and offered a deeper understanding of the underlying cell biology of B-ALL.
Back to topExtrapolating single cell B-cell states from bulk RNA-seq patient data reveals cell type behind resistance
The power of these computational methods carried over into the next part of their study, as the team used the single cell reference atlas of B-cell development and an algorithm called CIBERSORTx to perform cell-type deconvolution on bulk RNA-seq data from 1,986 B-ALL patient samples, which included 23 B-ALL molecular subtypes. Single cell RNA-seq profiles of the eight B-cell states previously identified were used to estimate the abundance of those B-cell states in bulk data. Importantly, this revealed that, across the patient samples, development was paused primarily at the pre-pro-B-like and pro-B-like stages (Figure 2).
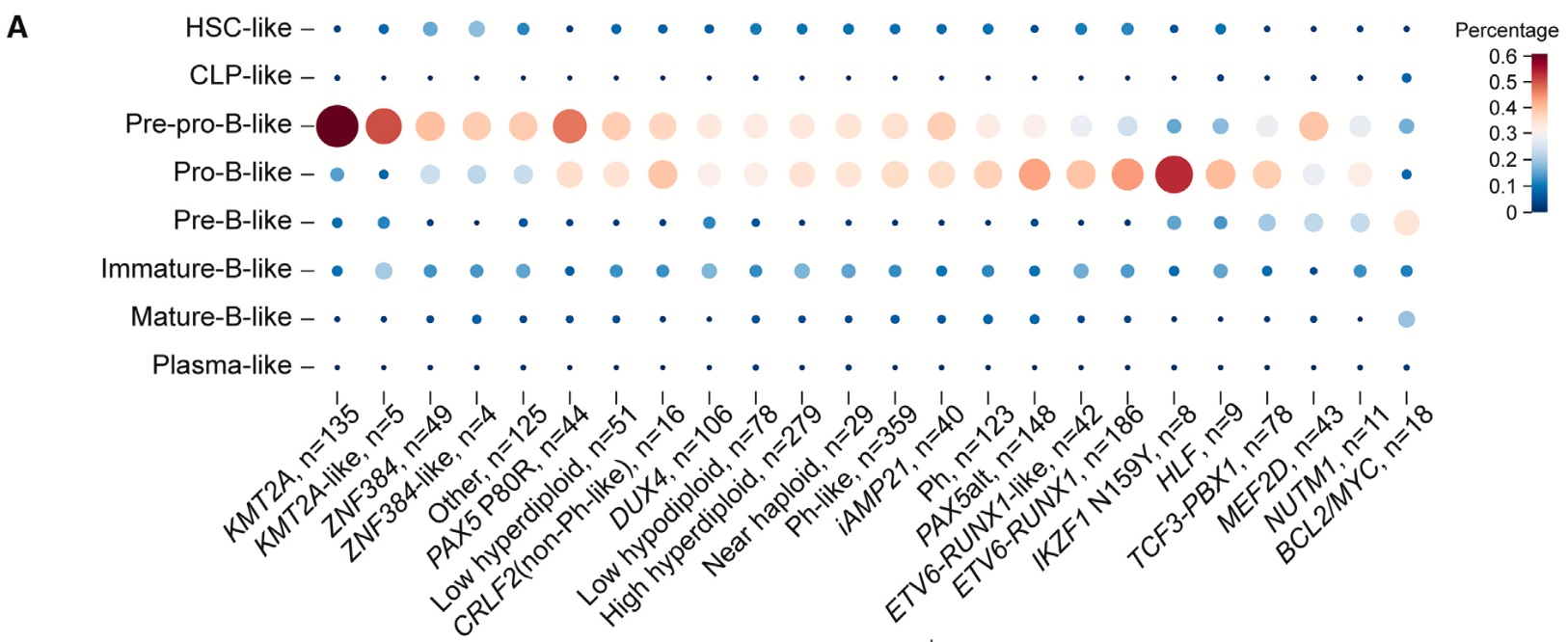
These results gave the team a foundation to build a hypothesis around the influence of B-cell developmental states on therapeutic response to asparaginase, a chemotherapeutic agent commonly used to treat leukemia since the 1960s. Despite a 94% overall survival rate, pediatric B-ALL patients who relapse or have asparaginase-resistant cancers show only a 30–50% survival rate (1), making this opportunity to refine the oncology community’s understanding of asparaginase resistance particularly crucial.
They analyzed bulk RNA-seq profiles of a new cohort of 203 B-ALL patients with SJARACNe and NetBID2 algorithms to infer the driver genes underlying asparaginase sensitivity and resistance, comparing the gene activity between responsive and resistant patient cases. This not only revealed eight asparaginase resistance genes and five sensitivity genes, but also demonstrated that the resistance genes had heightened activity in CLP and pre-pro-B developmental stages.
Using the single cell reference atlas to once again perform deconvolution of the bulk profiles in this cohort of patient samples also revealed that the asparaginase-resistant B-ALL cases had a higher proportion of pre-pro-B-like cells than asparaginase-sensitive samples. These findings suggested that the pre-pro-B-like stage may be the predominant cell population driving resistance to asparaginase treatment.
Back to topValidating resistant and sensitive B-cell states using Single Cell Multiome
These groundbreaking insights about the likely resistant B-cell development stage in B-ALL were made through innovative applications of single cell datasets and computation to bulk RNA-seq patient data. However, it was essential to observe these cellular dynamics at single cell resolution in B-ALL patient samples to further validate the hypothesis.
Using Chromium Single Cell Multiome ATAC + Gene Expression, the team performed joint chromatin accessibility and gene expression analysis in individual nuclei of primary blast cells from two asparaginase-sensitive and two resistant B-ALL cases (samples were taken prior to treatment). This joint analysis could allow them to better stratify B-cell states and confirm the relationship between B-cell developmental state and asparaginase response. They observed the exact results that they had inferred through previous methods: the two resistant samples consisted predominantly of a pre-pro-B-like population (84.9% and 91.9%), while the two sensitive samples were mostly pro-B-like cells (79.2% and 78.1%). These single cell findings added confidence to the hypothesis that pre-pro-B-like cancer cells were likely driving asparaginase resistance and pro-B-like cells were sensitive to asparaginase.
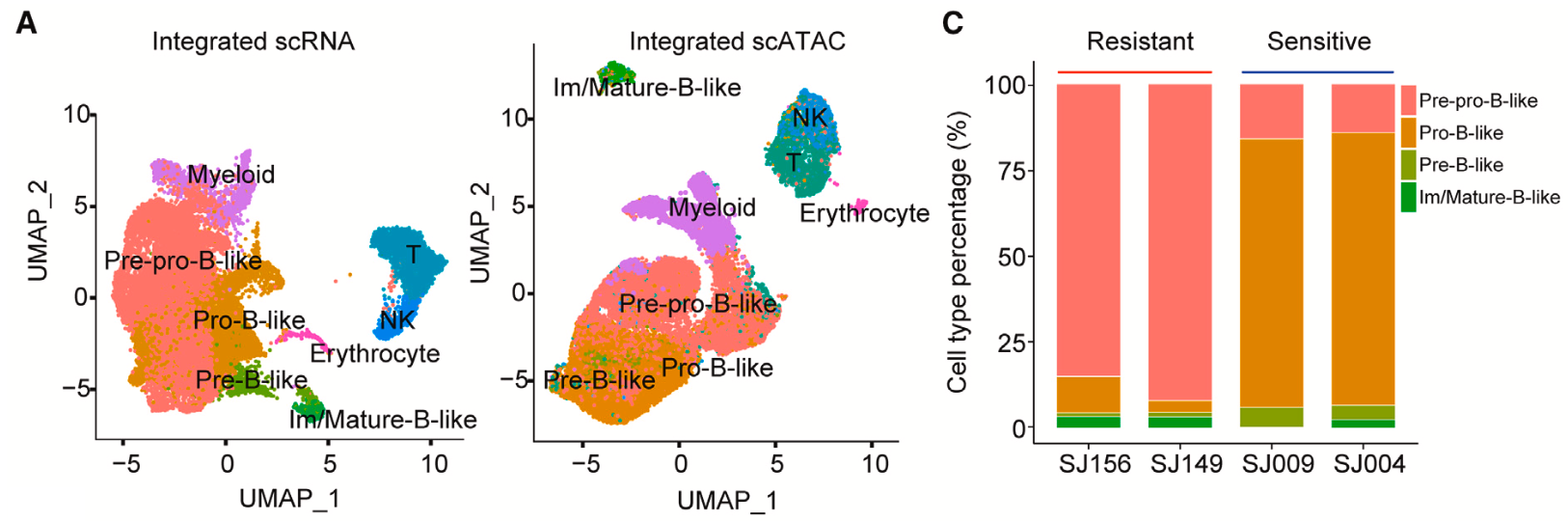
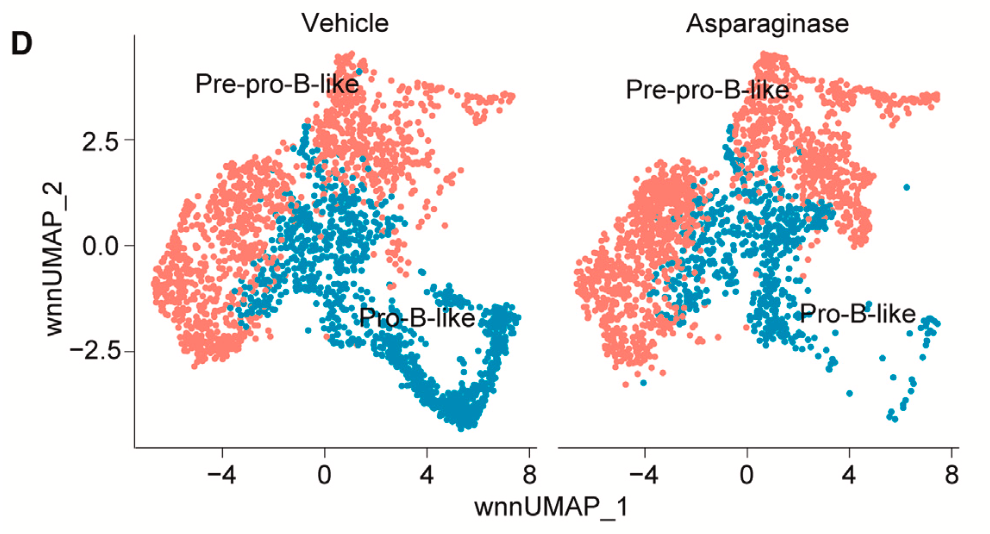
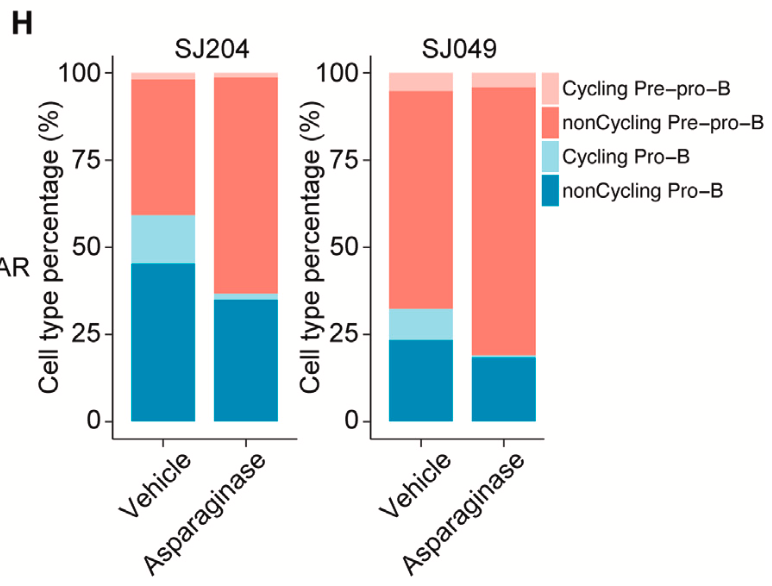
Could these same dynamics be seen in patient samples where there was intermediate or partial drug sensitivity? The research group performed Single Cell Multiome ATAC + Gene Expression analysis on two patient-derived xenograft (PDX) samples, which were established from patients that had this intermediate sensitivity. Culturing human leukemia cells isolated from mouse spleens with asparaginase for the treatment group, and a vehicle for the control group, the team noted a clear shift in leukemia cell populations as a result of treatment. There was a significant decrease in the pro-B-like subpopulation after asparaginase treatment and an increase in the pre-pro-B-like population (Figure 4). They also observed that asparaginase had particular efficacy towards cycling leukemia cells in the S-phase (when DNA is replicating), regardless of development state (Figure 5). These observations further confirmed the influence of B-cell development state and intratumoral heterogeneity on therapeutic efficacy and provided new details to the asparaginase mechanism of action.
BCL2 is a targetable protein in asparaginase-resistant B-ALL cells
What makes pre-pro-B-like cells resistant to asparaginase? And can that underlying biology be targeted to improve therapeutic response? To answer these questions, the team returned to their systems algorithms to identify the driver genes behind the pre-pro-B-like phenotype and possible protein targets. They used SJARACNe to define the interactome of pre-pro-B-like cells and then cross-examined driver genes to identify the top druggable candidates. This revealed BCL2 was a key driver gene in the pre-pro-B subpopulation, with peak activity in pre-pro-B cells and plasma cells, but reduced in the pro-B stage; moreover, its activity mirrored the pattern of the asparaginase-resistant gene signature across the phases of B-cell differentiation (Figure 6).
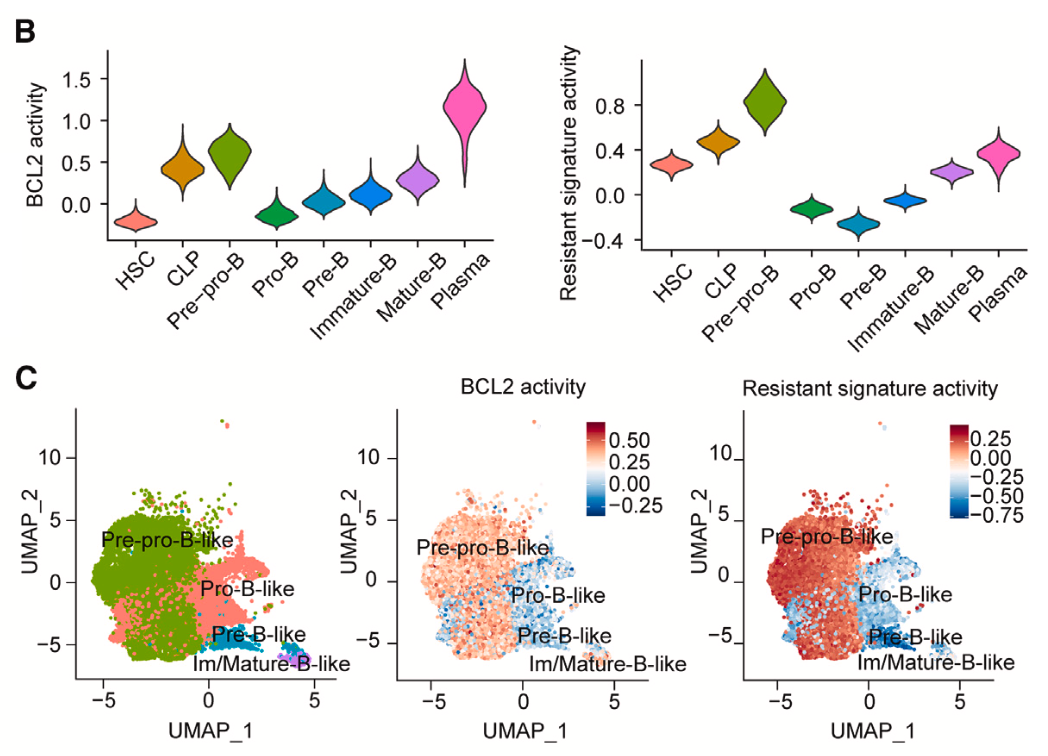
BCL2 (B-cell leukemia/lymphoma 2 protein) has well-characterized oncogenic effects: it works to block cell death through apoptosis, and is thus able to keep cancer cells from dying (3). Given the apparent heightened activity of BCL2 in asparaginase-resistant B-ALL cells, the research team reasoned that using an existing BCL2 antagonist (venetoclax) could overcome resistance and better kill leukemia cells.
Back to topFlex supports evaluation of combination therapy with asparaginase and venetoclax
The test of a good combination therapy is synergy. In a drug screening experiment assessing a multi-drug therapy, researchers look for a positive synergistic score that suggests the combined effect of the drugs is greater than the effect of each drug when used alone (4). 11 randomly selected B-ALL PDX samples were treated with the proposed combination asparaginase-venetoclax therapy, and, incredibly, the team saw significant positive synergistic scores in all samples.
To further tease out the underlying mechanism of action for asparaginase-venetoclax, the team performed single cell analysis on one PDX sample using Chromium Single Cell Gene Expression Flex. Primary B-ALL cells taken from the xenograft sample were treated for 24 hours with either asparaginase-venetoclax or the single drugs, while a subset of the sample represented an untreated control. After this treatment cycle, the team fixed the cells, then analyzed them with Flex. Though the authors do not comment on the reason for this choice of single cell assay, fixation can provide logistical benefits, allowing the team to essentially seal the sample in the state it was in after 24 hours of treatment (to give the most accurate readout of the drug effects) and then perform the assay on a more flexible schedule. Additionally, the high gene expression sensitivity of this probe-based approach could allow the team to obtain the most accurate results for low-expressed genes.
Flex single cell data confirmed their hypothesis regarding asparaginase activity: the sample subset treated exclusively with asparaginase showed a large reduction of the pro-B-like cancer cell state (43.15% down to 8.39%). And, though the combined therapy showed an overall positive effect to reduce blast populations, the assessment of venetoclax alone revealed that it had a small impact on pre-pro-B-like cell state (56.85% down to 54.63%).

These findings suggested that venetoclax was likely working to increase the efficacy of asparaginase, overcoming barriers the latter drug could not accomplish alone. Notably, through pathway enrichment analysis of single cell RNA-seq data from the drug-treated and control cells, the team observed that asparaginase downregulated mTOR signaling, priming B cells for apoptosis and perhaps making them dependent on BCL2 to prevent cell death. Thus, asparaginase activity could sensitize B cells to venetoclax activity to block BCL2, driving their synergistic effect.
To better understand if the combination therapy was more effective overall and could improve upon current treatment regimens in areas such as progression-free survival and tumor burden, the team turned to in vivo mouse tumor models. They treated a cohort of mice with the asparaginase-venetoclax combination therapy and observed a significant reduction of peripheral blood leukemia burden after two weeks, which was a better result than either drug alone. Mice treated with the combination therapy also showed prolonged survival compared to control or single-drug groups. These positive results suggested that the combination therapy could be valuable to better treat high-risk B-ALL.
Back to topFrom first principles–driven data exploration to a novel combination therapy
This study modeled a powerful approach to advance our knowledge of disease biology and build better therapies. Deeply characterizing the healthy biological system (in this case, B-cell developmental states) established a data resource that enabled further exploration of hypotheses about what went wrong in the context of disease (B-cell cancer). Innovative algorithms unlocked the full potential of both large-scale healthy human datasets and data from precious patient samples, revealing patterns that pointed to the cell types and driver genes behind disease biology and therapeutic resistance. Taking these findings one step further, researchers used data-driven logic to explore new possibilities for therapeutic improvements.
We’re deeply impressed and inspired by this study for not only the robust approach to drug discovery that it exemplifies, but also for how it cycled between clinical and pre-clinical settings to maximize and extend truly precious patient samples and data. The samples used for this study included pediatric cases from St. Jude Children’s Research Hospital (sampled as part of a clinical trial, NCT03117751) and adult samples from MD Anderson Cancer Center, the University of Chicago, and two additional clinical trials (NCT02003222 and NCT03150693). By applying single cell tools that offered multiomic readouts and ensured the highest degree of gene expression sensitivity, computational tools that could derive deeper insights from single cell data, and xenograft models that expanded human samples for downstream cell culture, the team demonstrated what’s possible with the full range of powerful resources and methods available to the scientific community.
Learn more by reading the publication. And discover the single cell and computational tools they used:
- Chromium Single Cell Multiome ATAC + Gene Expression
- Chromium Single Cell Gene Expression Flex
- SJARACNe
- NetBID2
- CIBERSORTx
References:
- St. Jude Children’s Research Hospital. Targeting vulnerability in B-cell development leads to novel drug combination for leukemia. News Release. April 8, 2024.
- Huang X, et al. Single-cell systems pharmacology identifies development-driven drug response and combination therapy in B cell acute lymphoblastic leukemia. Cancer Cell 42: 552–567.e6 (2024). doi: 10.1016/j.ccell.2024.03.003
- https://www.cancer.gov/publications/dictionaries/cancer-terms/def/bcl2
- https://www.cancer.gov/publications/dictionaries/cancer-terms/def/synergistic
About the author:

Blog contributor: This article has been reviewed for scientific accuracy by Angela Churchill, PhD.
