Xenium Onboard Analysis
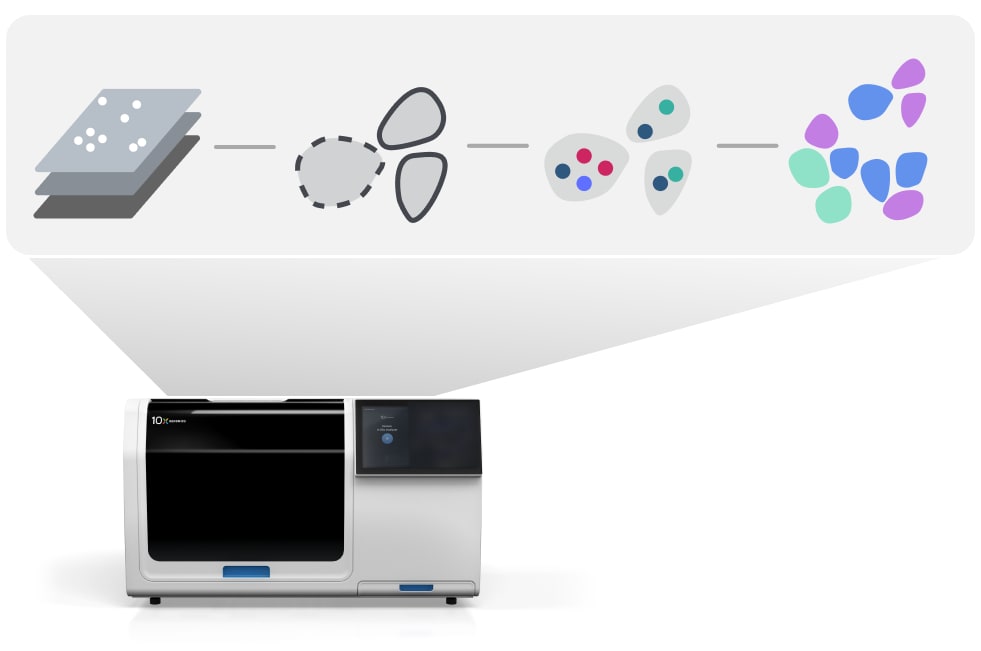
What is Xenium Onboard Analysis?
The Xenium In Situ software suite is a set of software applications for analyzing and visualizing in situ gene expression data produced by the Xenium Analyzer. The Xenium Onboard Analysis pipeline enables simultaneous on-instrument data collection and analysis.
Analysis
Estimate and Monitor Xenium Analyzer Instrument Run Time
A table of Xenium onboard analysis pipeline output files.
Overview of all Xenium onboard analysis pipeline output files.
Overview of the interactive Xenium analysis summary output file metrics and plots.
Troubleshooting guide for the Xenium analysis summary alerts.
Descriptions for protein data-specific outputs.
Storing Xenium raw data for reanalysis or grant requirements.
Algorithms
View the latest updates to Xenium Onboard Analysis algorithms.
Release Notes
View the latest updates to Xenium Onboard Analysis.
Xenium Analyzer
Powerful in situ profiling platform using curated panels, powerful visualization software, and easy-to-follow workflow.
Xenium Explorer
Visualize data from Xenium In Situ solutions.
Xenium Ranger
Reanalyze data from Xenium In Situ solutions.
Q&A
Browse Q&As from the 10x Genomics support team.
Analysis Guides
A collection of introductions, tutorials, and blogs for data analysis beyond 10x Genomics software.