Many experiments involve generating data from multiple samples. Depending on the experimental design, these could be multiple capture areas from consecutive sections of the same tissue block, or samples from different biological conditions of the same tissue. The spaceranger aggr pipeline can be used to aggregate samples into a single feature-barcode matrix.
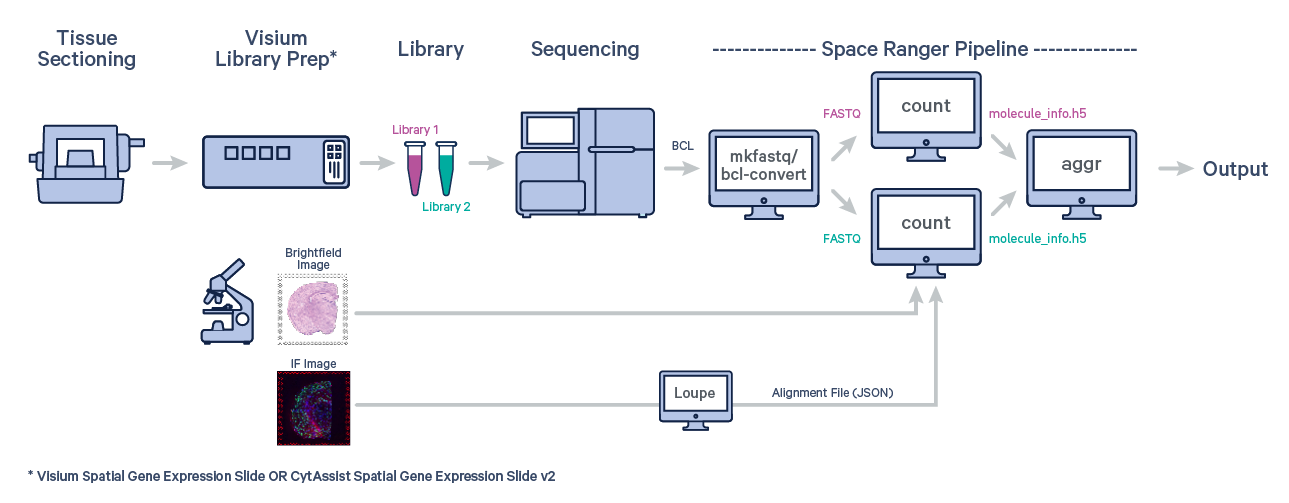
The first step is to run spaceranger count on each individual capture area from the Visium slide. For example, suppose you ran three instances of spaceranger count:
cd /opt/runs spaceranger count --id=LV123 ... # wait for pipeline to finish spaceranger count --id=LB456 ... # wait for pipeline to finish spaceranger count --id=LP789 ... # wait for pipeline to finish
Next, you need to create an aggregation CSV file.
Create a CSV file with a header line containing the following columns:
| Column Name | Description |
|---|---|
library_id | Required. Unique identifier for this input capture area. This will be used for labeling purposes only; it doesn't need to match any previous ID you have assigned to the capture area. |
molecule_h5 | Required. Path to the molecule_h5 file produced by spaceranger count. For example, if you processed your capture area by calling spaceranger count --id=ID in some directory DIR, this path would be /DIR/ID/outs/molecule_info.h5. |
cloupe_file | Required. Path to the cloupe.cloupe file produced by spaceranger count. For example, if you processed your capture area by calling spaceranger count --id=ID in some directory DIR, this path would be /DIR/ID/outs/cloupe.cloupe. |
spatial_folder | Optional. Path to the spatial file produced by spaceranger count. For example, if you processed your capture area by calling spaceranger count --id=ID in some directory DIR, this path would be /DIR/ID/outs/spatial. Note that not specifying the spatial_folder column will lead to the aggregated tissue positions list (aggr_tissue_positions.csv) and spatial folder containing spatial images and scalefactors (spatial) to be omitted from the outs folder generated from thespaceranger aggr run. |
The CSV can be made either in a text editor or created in Excel and exported to CSV. Continuing the example from the previous section, the saved CSV would look like this:
library_id,molecule_h5,cloupe_file,spatial_folder LV123,/opt/runs/LV123/outs/molecule_info.h5,/opt/runs/LV123/outs/cloupe.cloupe,/opt/runs/LV123/outs/spatial LB456,/opt/runs/LB456/outs/molecule_info.h5,/opt/runs/LB456/outs/cloupe.cloupe,/opt/runs/LB456/outs/spatial LP789,/opt/runs/LP789/outs/molecule_info.h5,/opt/runs/LP789/outs/cloupe.cloupe,/opt/runs/LP789/outs/spatial
In addition to the CSV columns expected by spaceranger aggr, you may optionally supply additional columns containing library meta-data (e.g., lab or sample origin). These custom library annotations do not affect the analysis pipeline but can be visualized downstream in the Loupe Browser (see below). Note that unlike other CSV inputs to Space Ranger, these custom columns may contain characters outside the ASCII range (e.g., non-Latin characters).
It is possible to assign categories and values to individual samples prior to the spaceranger aggr run by adding additional columns to the aggregation CSV. These category assignments propagate into Loupe Browser, where you can view them, and determine genes that drive differences between samples. For example, the following spreadsheet was used to aggregate the bundled Loupe tutorial dataset:
| library_id | molecule_h5 | cloupe_file | AMLStatus |
|---|---|---|---|
| AMLNormal1 | /path/to/AMLNormal1/outs/molecule_info.h5 | /path/to/AMLNormal1/outs/cloupe.cloupe | Normal |
| AMLNormal2 | /path/to/AMLNormal2/outs/molecule_info.h5 | /path/to/AMLNormal2/outs/cloupe.cloupe | Normal |
| AMLPatient | /path/to/AMLPatient/outs/molecule_info.h5 | /path/to/AMLPatient/outs/cloupe.cloupe | Patient |
Any columns in addition to library_id, molecule_h5, cloupe_file and spatial_folder will be converted into categories, and the spots in each sample will be assigned to one of the values in that category.
spaceranger aggr is not designed to perform batch correction for removal of technical artifacts due to differences across assays. While Cell Ranger's chemistry batch correction is enabled in the pipeline, it is not supported. To use this option, include the following column in your aggregation CSV file:
- batch: Optional. Unique identifier for the batch that this library belongs to. Libraries with the same batch identifier are considered to be in the same batch.
10x Genomics does not recommend combining Visium data from fundamentally different treatments such as different staining protocols (e.g., immunofluorescence vs H&E stained tissue sections), different tissue storage conditions (e.g., Fresh Frozen vs FFPE tissue sections), different library preparation protocols (e.g., short cDNA vs long cDNA), or other variations in the assay preparation.
Run spaceranger aggr --help for a full list or refer to the Command Line Arguments Reference page for details. After specifying these input arguments, run spaceranger aggr:
cd /opt/runs
spaceranger aggr --id=AGG123 \
--csv=AGG123_libraries.csv \
--normalize=mapped
The pipeline will begin to run, creating a new folder named with the aggregation ID you specified (e.g. /opt/runs/AGG123) for its output. If this folder already exists, spaceranger will assume it is an existing pipestance and attempt to resume running it.
The spaceranger aggr pipeline generates output files that contain all of the data from the individual input jobs, aggregated into single output files, for convenient multi-sample analysis.
When combining multiple capture areas, the barcode sequences for each channel are distinguished by a capture area suffix appended to the barcode sequence.
By default, the reads from each capture area are subsampled such that all capture areas have the same effective sequencing depth, measured in terms of reads that are confidently mapped to the transcriptome or assigned to the feature IDs per spot. However, it is possible to change the depth normalization mode.
To aggregate libraries with spaceranger aggr, slide versions and chemistries must match. Aggregating Visium HD data is not supported.
Samples run with an antibody feature_reference.csv in each individual capture area can only be aggregated with other samples that were ran with spaceranger count using the same feature reference. Re-run spaceranger count with a common feature reference (see --no-libraries) to allow aggregation.
When combining data from multiple capture areas, the spaceranger aggr pipeline automatically equalizes the read depth between groups before merging, which is the recommended approach in order to avoid the batch effect introduced by sequencing depth. The none option may be appropriate if you want to retain all data.
There are two normalization modes:
| Argument | Description |
|---|---|
--normalize=mapped | Default. Subsample reads from higher-depth capture areas until they all have, on average, an equal number of reads per tissue covered spot that are confidently mapped to the transcriptome. |
--normalize=none | Optional. Do not normalize at all. |
Each capture area is a physically distinct partition on a Visium slide printed with the same set of barcodes. To uniquely identify barcodes when aggregating multiple libraries, Space Ranger appends a suffix to the barcode in the feature-barcode matrix. For example, AAACAACGAATAGTTC-1 and AAACAACGAATAGTTC-2 are distinct spot barcodes from different capture areas, despite having the same barcode nucleotide sequence. The numbering reflects the order that the capture areas were provided in the aggregation CSV.