How does single cell RNA-seq work

Editor’s note: The purpose of this piece is to introduce how scRNA-seq works by following the journey of an RNA molecule through the wet-lab workflow. Some steps have been simplified for clarity. If you are looking to understand the benefits of single cell, we recommend you check out our beginner's guide to single cell biology. For comprehensive technical details, we recommend consulting the user guides specific to the 10x Genomics assay of interest.
The journey from mRNA to sequencing-ready libraries
One of the first single cell RNA sequencing (scRNA-seq) studies was published in 2009 by Tang et al., marking a significant advancement in transcriptomics (1). As the name implies, RNA molecules are the stars of scRNA-seq experiments. When you stop to think about it, the process of transforming RNA to cDNA and then into digital data is quite remarkable.
So, how does scRNA-seq work? Let’s explore the answer by considering the “life” of an RNA molecule within the scRNA-seq experiment. RNA molecules experience two “lives” in this context. The first comprises the “physical” journey that starts with messenger RNA (mRNA) isolation from its cell of origin and ends with the generation of the sequencing-ready library. Upon sequencing, the RNA then takes on a “digital” journey that is ultimately transformed into your single cell data.
This two-part blog walks through those two “lives,” exploring the wet-lab scRNA-seq workflow (Part 1) and digital analysis (Part 2), in more detail—providing a whole new appreciation for the awesome power of this method. Keep reading to explore the “physical” life of RNA molecules, where we break down the wet-lab scRNA-seq workflow into four key milestones:

What is the difference between scRNA-seq and RNA-seq?
Both bulk RNA sequencing (RNA-seq) and scRNA-seq assess gene expression. However, bulk RNA-seq involves extracting, pooling, and sequencing RNA from numerous cell types within a sample (2). Thus, this approach only provides an ensemble readout. This averaging obscures rare cell types or subtle gene expression differences among individual cell types in the sample.
Chronicling the “physical” life of an mRNA molecule in the scRNA-seq workflow
Sample dissociation
The journey from mRNA to sequencing library begins inside of your sample. These days, a variety of sample types can be used as inputs for single cell experiments. Our Chromium Single Cell portfolio, for example, features assays compatible with fresh, frozen, and fixed tissue samples—even FFPE.
It’s important to remember that “garbage in, garbage out” is just as relevant for biological experiments as it is in computer sciences. That’s why generating high-quality single cell—or in some cases single nuclei—suspensions is a critical step in your experiment. The approach you take to create this suspension is highly dependent on your sample type and the single cell assay you will be performing. We recommend consulting the support documentation available to you before tackling this step.
Spotlight on Flex Gene Expression
Our Flex Gene Expression assay provides sensitive whole transcriptome, protein-coding gene coverage for human and mouse samples, including those with low-quality RNA. Its unique workflow allows quick and easy sample fixation after collection, enabling safe storage and transport without affecting RNA integrity or data quality. When ready, fixed samples are permeabilized and incubated with the Flex gene expression panel targeting 18,000+ protein-coding genes, which then become the input for cell partitioning.
Cell partitioning and barcoding
Getting your suspension sorted into single cells and the mRNA molecules barcoded is key to a successful single cell experiment. In 10x assays, Chromium X series instruments use advanced microfluidics technology to automate these crucial steps. Think of this instrument support as having a super-efficient, reliable lab assistant that ensures top-notch, reproducible results every single time.
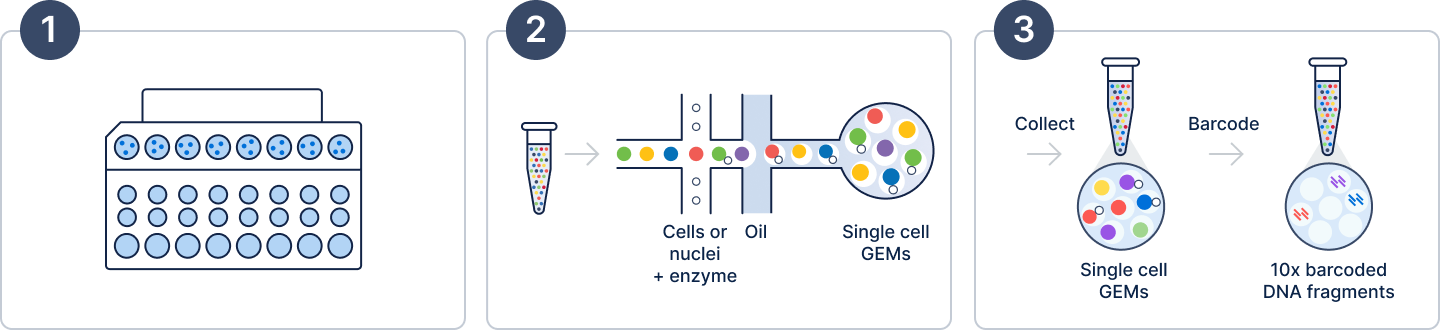
The process begins by loading your suspension of single cells or nuclei and the reagents for your selected assay—including lysis buffer, oligonucleotides, Gel Beads, and enzymes—onto a microfluidic chip (Figure 2, Step 1). Next, within the Chromium X series instrument, cells move through the channels at a limiting dilution to generate tens of thousands of Gel Beads-in-emulsions, which we call GEMs, in just minutes (Figure 2, Step 2).
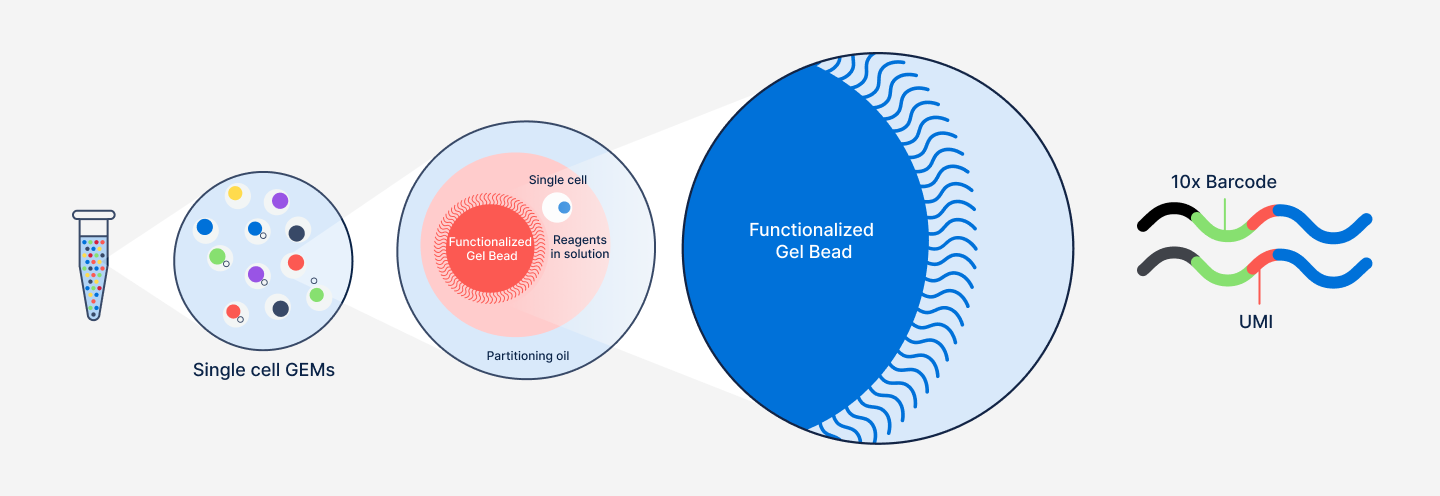
GEMs are small but mighty, each one acting as an individual reaction droplet (Figure 3). Immediately following GEM generation, the Gel Bead is dissolved, releasing the barcoded Gel Bead primers, and the cell is lysed, releasing the mRNA transcripts.
At this point, an incubation step allows the creation of barcoded fragments that will become the inputs for library generation (Figure 2, Step 3). Importantly, all fragments from the same cell or nucleus share a common 10x Barcode, allowing them to be traced back to their cell of origin. It is worth noting that each fragment also contains a unique molecular identifier (UMI) sequence. These sequences are different for every Gel Bead primer, and help ensure accurate “counting” of transcript representation in individual cells. We'll come back to these more in-depth when discussing data analysis in Part 2.
Spotlight on Universal Gene Expression
Our Universal 3’ and 5’ Gene Expression assays deliver robust and reliable single cell profiling suitable for samples originating from a wide variety of species. These assays create whole transcriptome single cell libraries by capturing the 3’ or 5’ ends of mRNA transcripts following GEM generation. This scRNA-seq chemistry approach makes them ideal for gathering broad information such as isoforms, single nucleotide variants, long non-coding RNAs, and more.
A closer look at barcoding
Many steps that take place within these droplets following the release of the Gel Bead primers and mRNAs. The steps, detailed briefly below, vary depending on whether you are using a Flex or Universal Gene Expression assay.
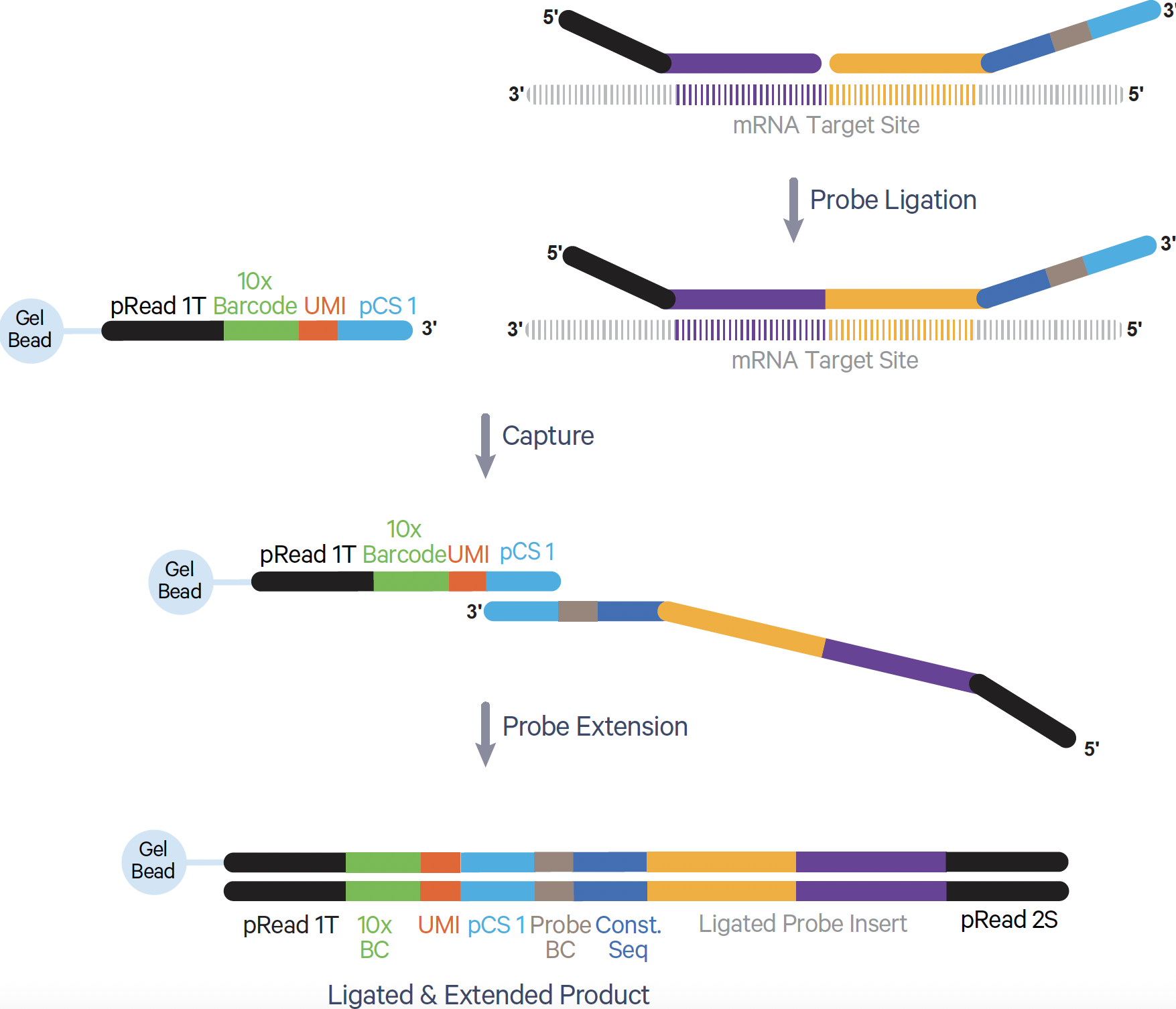
- In the Flex assay, probes hybridize to protein-coding mRNA targets in fixed and permeabilized cells. Following GEM generation, a ligation reaction closes the gap between the probe pairs bound to a given mRNA target (Figure 4). Gel Bead primers then bind, and a polymerase extends the probe to include the UMI and 10x GEM Barcode as well as a partial Read 1T (pRead 1T) and partial Read 2S (pRead 2S) sequence—these sequences will come in handy during library preparation.
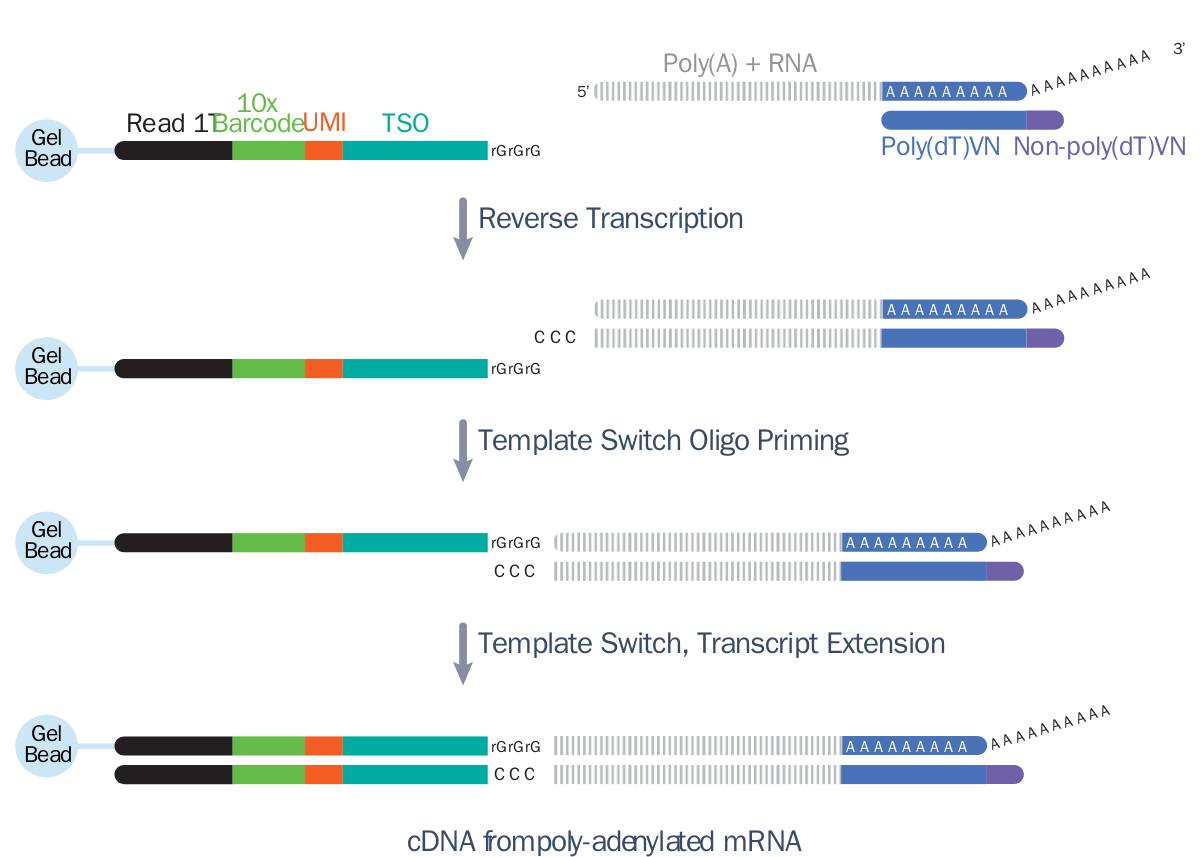
- With the Universal 3' assay, the familiar method of reverse transcription (RT) creates the barcoded fragments following GEM generation (Figure 5). Briefly, the poly(dT) sequence on the Gel Bead primer binds to mRNA transcripts with a poly(A) tail. Incubation with the appropriate RT enzymes synthesizes a complementary DNA (cDNA) molecule containing a 10x Barcode, a pRead 1T sequence, and a template switch oligo (TSO).
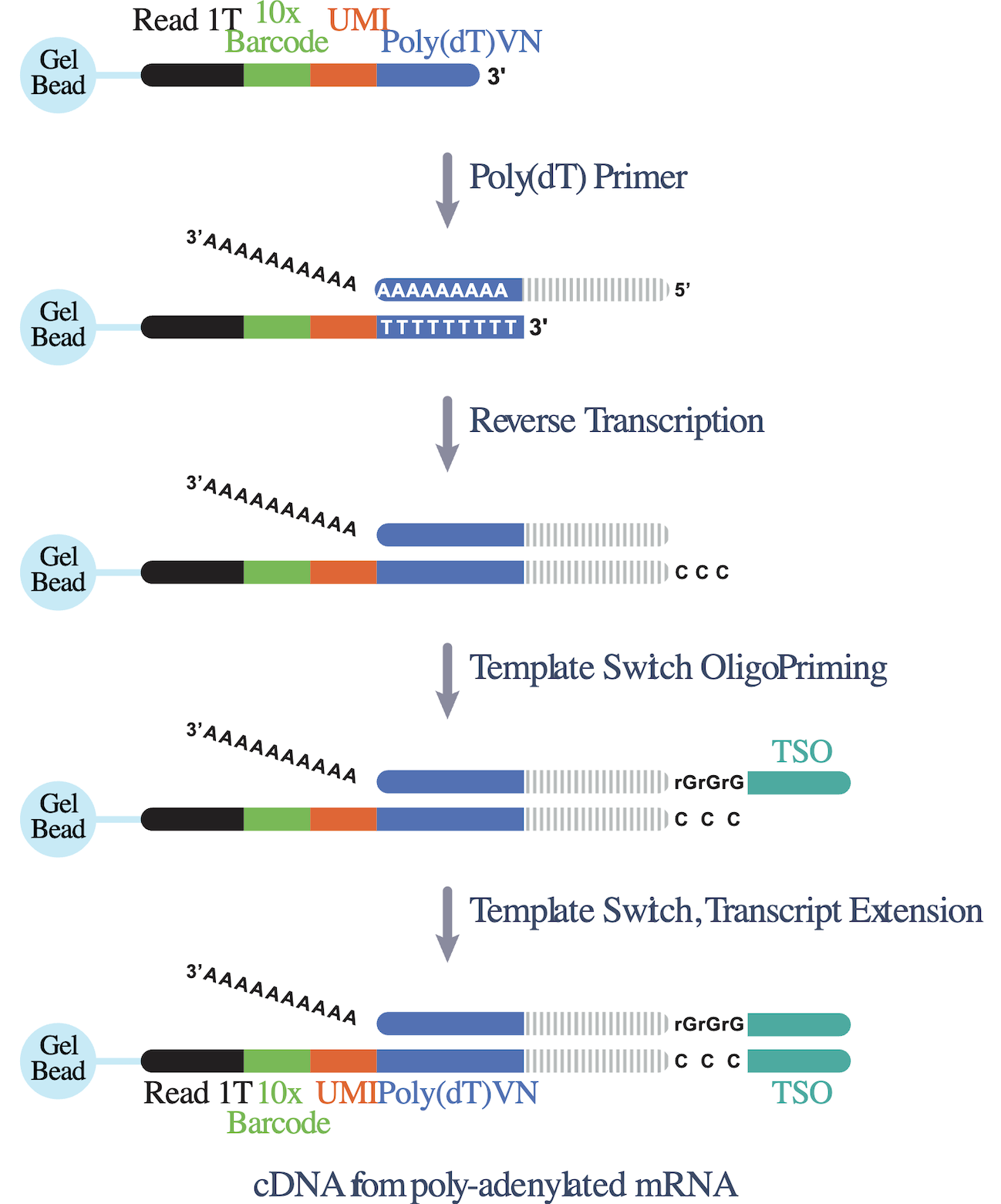
- The Universal 5' assay also leverages RT to generate barcoded fragments (Figure 6). However, since this assay profiles the 5' end of mRNA transcripts, it has some unique steps. In this assay, a poly(dT) oligo in the reaction mix binds to the poly(A) tail of an mRNA transcript, and an RT reaction makes the cDNA strand. A TSO located on the Gel Bead primer allows the cDNA strand to become the template for transcript extension, resulting in the incorporation of a 10x Barcode, a UMI, and a pRead 1T onto this cDNA strand.
Before moving on to the next step, let’s take a moment to revisit our friend the mRNA molecule. We started this post highlighting mRNA as the star of an scRNA-seq experiment. However, you may have noticed that, by the end of the GEM formation and barcoding step, something curious happened.
Our mRNA molecule has passed the torch. Its essence now lives on in the final outputs for the Flex and Universal assays. These outputs are not mRNA transcripts but a ligated, extended nucleotide product and a cDNA, respectively.

More than gene expression: Multiomic readouts
One of the advantages of the Chromium portfolio is that it gives you the flexibility to tackle a wide range of analytes and applications alongside gene expression. Measure protein expression, perform comprehensive T- and B-cell receptor sequencing, assess CRISPR perturbations, and map regions of open chromatin, all at single cell resolution.
Amplification
We now have barcoded molecules, but we aren’t quite ready to sequence just yet. One of the primary reasons for this is that single cells contain relatively small amounts of transcripts. We therefore need to amplify these barcoded molecules to be able to detect them. Again, this step looks slightly different depending on which assay is being used.
- For the Flex assay, this step is called Pre-amplification. First, the GEMs are broken up to release the ligated, barcoded product. Then, a polymerase chain reaction (PCR) is used to make many copies of the ligated products.
- In the Universal 3' and 5’ assays, the GEMs are first broken up to release the barcoded cDNA products. A purification step is used to obtain the first-strand cDNAs. Next, PCR is used for cDNA amplification.
Sequencing library preparation
We are nearing the end of our physical journey. However, the amplified products must first undergo sample indexing. In 10x assays, the Sample Index PCR step serves two purposes. First, it adds sample index sequences that allow you to mix multiple samples for a single sequencing run—a standard practice for most sequencers. Second, it adds priming sites required to perform next-generation sequencing. For this blog, we will focus on the importance of the sequencing priming sites.
A quick note before discussing library preparation details for our assays. It is worth mentioning that there are multiple compatible sequencers—including Illumina, Ultima Genomics, Oxford Nanopore, and more—depending on your chosen assay. Consult the appropriate product documentation to get more details.
- In the Flex assay, the Sample Index PCR is performed on amplified ligated products that have undergone a cleanup step. This PCR is where the pRead 1T and pRead 2S, introduced during barcoding, come back into play. During the PCR step, these are recognized by the PCR primers and allow the P5 and P7 sequences to be added.
- The Universal 3' and 5’ assays require the amplified cDNA products to undergo multiple steps. The two that we will discuss here are enzymatic fragmentation and adapter ligation. Enzymatic fragmentation helps create shorter fragments that are better suited for sequencing. Adapter ligation allows the Read 2T sequence to be added. Both the pRead 1T, introduced during barcoding, and this 2T sequence are used in a PCR step to add the P5 and P7 sequences.
With this, we have sequencing-ready libraries and have completed the last major milestone in the “physical” life of the mRNA.
The end of one life, the beginning of another
From its origins inside a single cell to its transformation into a sequencing-ready library, the physical journey of mRNA molecules in scRNA-seq is as fascinating as it is intricate. Each step along the way has played a critical role in preserving the biological information encoded in the original mRNA transcripts.
However, this chronicle is far from over. In Part 2, we’ll continue the journey and explore mRNA’s “digital” life. That is, the digital transformation of the transcript sequence, via sequencing, and computational analysis that reveals the gene expression information we are after.
References:
- Tang F, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 6: 377–82. doi: 10.1038/nmeth.1315.
- “Choosing Genomics Tools.” Chapter 13 Single-Cell RNA-Seq, The Fred Hutch Data Science Lab, hutchdatascience.org/Choosing_Genomics_Tools. Accessed 2 July 2025.
About the author:
