Ask the 10x-perts: 4 reasons to upgrade your Cell Ranger ATAC and ARC software

Ready to make a good thing better? Cell Ranger ATAC and ARC software have just received some major improvements! Update to Cell Ranger ATAC v2 and Cell Ranger ARC v2 to see how these powerful tools for computational analysis of single cell open chromatin and transcriptional data have gotten even better.
Designed to complement the Chromium Single Cell ATAC assay, Cell Ranger ATAC analysis pipelines allow you to assess chromatin accessibility at the single cell level, providing insights into cell types and states and deeper understanding of gene regulatory mechanisms. Building on this capability, Cell Ranger ARC, part of the Chromium Single Cell Multiome ATAC + Gene Expression solution, not only infers regions of open chromatin in single cells but also links them to gene expression data from those same cells.
Wondering how Cell Ranger ATAC and ARC could get even better? We asked three 10x-perts more about the software improvements and why they think it’s worth upgrading to v2.
1. Improved peak calling
According to Brett Olsen, Staff Computational Biologist at 10x Genomics, Cell Ranger ATAC v2 and Cell Ranger ARC v2 include “some substantial improvements to how we identify accessible regions of chromatin, or peaks, in our ATAC samples.” These peaks are a key component of many downstream analyses using Cell Ranger ATAC, including cell clustering, cell-type identification, and peak motif identification. The same is true for Cell Ranger ARC, which also determines feature linkages between ATAC peaks and gene expression.

Olsen explains that in the previous versions of the software, peaks were called using a common background noise threshold across the whole genome. This often yielded very long peaks, which, Olsen tells us, “tended to occur in regions of high local background and merged signal from multiple true peaks into a single peak.” These long peaks often obscured differences in cellular behavior between merged peaks and made peak motif identification difficult.
Now, in v2, this initial threshold is instead used to identify candidate regions—areas of the genome that may include peaks. The signal inside each candidate region is then examined for potential peaks based on local background estimates. This method provides a more accurate estimation of local background around potential peaks, ensuring consistent peak calling in different genomic regions with different levels of background noise.
However, there’s a caveat. Olsen says, “We found that simple local background tests can hide peaks in regions with many peaks: neighboring strong peaks can skew the local background estimate too high, preventing nearby smaller peaks from being accurately identified.” To solve this problem, local backgrounds are estimated while excluding any potential peaks in the region
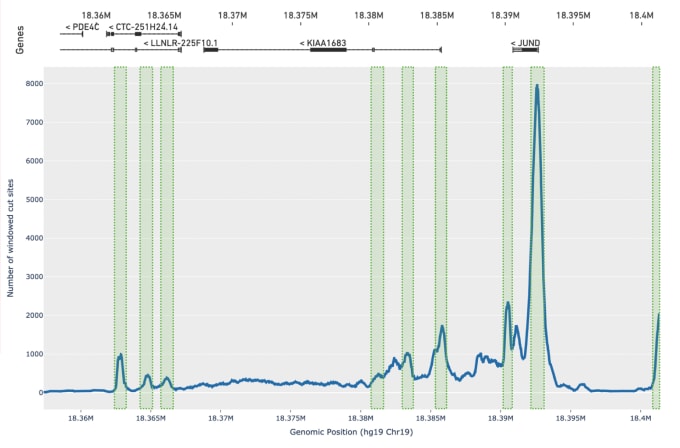
Olsen tells us this improved peak calling method has added benefits. In addition to improving the accuracy of peak calls, it also provides more robust results from samples with very low-quality data, which is often due to poor chromatin structure and enzyme targeting. He says, “While our previous caller had difficulty calling any peaks on these types of samples, we now identify an order of magnitude more peaks, enough to do basic cell clustering and cell identification even with very low-quality input data.”
2. Increased sensitivity
Refined peaks aren't the only performance improvements for Cell Ranger ATAC v2 and ARC v2. How does a 25% increase in ATAC median fragments per cell sound? An update to our duplicate marking algorithm is making this possible. Now, read pairs are considered duplicates if they share the same starting position, end position, and cell barcode—previously, only the start and end were used. This reduces the occurrence of false positives when identifying duplicates.
And, the end result? More ATAC median fragments per cell and increased sensitivity for your experiments.
3. Faster runtime
Based on Staff Computational Biologist Vijay Kumar’s calculations, Cell Ranger ATAC v2 and ARC v2 run about four times faster and require approximately 50% less disk space than v1. This one’s pretty simple, right? The faster your runtime, the more you save—in time and resources.

But while the advantage here is clear, you might be wondering how it works. Kumar explains that the team began by studying the existing pipelines and identifying the critical path. This turned out to involve two main factors. First, a series of sequential read processing stages. Then, an ATAC differential accessibility computation. Kumar and his team focused on finding ways to speed up these steps, identifying key points where processing could be more efficient. For example, they rewrote the ATAC read processing pipeline in Rust, which, as a compiled language, is naturally faster than the previous python code. They made a similar change to the differential accessibility step, rewriting the algorithm that computes enrichment of chromatin accessibility in Rust. They also changed the parallelization scheme, or the steps of the code that could be run in tandem. As Kumar explains, they tested two different schemes in Rust and found that their new approach “performed the best and resulted in a 5x speedup over the python version.” Not too shabby!
4. Multi-sample analysis in Cell Ranger ARC
That’s right. Cell Ranger ARC v2 is getting a major upgrade. With the addition of two new pipelines, aggr and reanalyze, users can now analyze data from multiple samples together.
Andrew Gottscho, Senior Scientific Writer, gave us some details on what this means for Single Cell Multiome ATAC + Gene Expression users. He explains, “The new aggr (shorthand for aggregate) pipeline in Cell Ranger ARC v2 provides a flexible way to combine data from multiple GEM wells in a single feature barcode matrix and other output files.” This allows easy visualization and analysis of large nuclei counts (>100,000 nuclei). He continues, “Meanwhile, the new reanalyze pipeline enables users to quickly tweak the details of their analysis, such as honing in on a specific set of genomic features (peaks) or a subset of cells, producing new outputs without having to process raw sequencing data from the beginning.” This gives users the freedom to focus more on deciphering the complex interactions between chromatin accessibility and gene expression. According to Gottscho, identifying the causal relationships between epigenetics and transcriptional output is where the “important breakthroughs in personalized medicine are waiting to be discovered, and that’s why our customers asked us to develop the product in the first place.”

Ready to upgrade your software? Update to v2 now:
Download Cell Ranger ATAC v2 →
Check out the 10x Genomics blog to learn more about Chromium Single Cell Multiome ATAC + Gene Expression:
- Find out how Single Cell Multiome ATAC + Gene Expression lets you simultaneously capture the transcriptome and the epigenome in the same single cells. Read more →
- Get the answers to your questions about Single Cell Multiome ATAC + Gene Expression. Explore FAQs →
- Optimize your sample prep techniques for single cell multiomics. Find out how →
- Discover how scientists leveraged both single cell ATAC and gene expression data to better understand the dysregulation underlying Alzheimer’s and Parkinson’s disease. Read now →
Download our recent data spotlight to see how researchers used Chromium Single Cell Multiome ATAC + Gene Expression to identify a tumor–specific gene regulatory network in human B-cell lymphoma.
Learn more about our turnkey software tools for data analysis and visualization, download datasets, and more on the 10x Genomics support site.
This blog was co-authored by Brett Olsen, Staff Computational Biologist, Vijay Kumar, Staff Computational Biologist, and Andrew Gottscho, Senior Scientific Writer at 10x Genomics.