By default, genes are categorized by panel design source in the Transcripts layer (i.e., "Predesigned", "Custom", or "Boosted"). Click on the Search for genes to visualize bar to select or type in specific gene names or organize genes by custom group labels.
In Xenium Explorer v3.1 and later, click the toggle (±) next to the search bar to collapse (default) or expand gene groups.
When zoomed in and with tooltips shown (Settings > Show Tooltips), you will see ID, location, and Q-Score for individual transcripts as you move the cursor over each transcript point.
Click the link below to learn how to explore, select, and export transcript statistics.
Transcript counts can be shown as points or as a density map. Starting in Xenium Explorer v3.0, the default maximum number of transcript points to load in the viewport is set to 5 million.
If a dataset exceeds this maximum value, Xenium Explorer auto-switches to density map view. You can select a subset of genes to see transcripts as points and with scaled transcripts view. The maximum value can be adjusted higher, however app performance may be affected.
When displayed as a density map, the cumulative density of all checked genes will be visible in the viewing area. The density map opacity, bin size, and scale threshold can be adjusted with the sliders (click arrow above maximum threshold value to reset). There are several density map palette options as well.
When colored by the density map, the lowest transcript density bin maps to the lower end of the palette (i.e., Inferno=black, Viridis=purple), while the highest transcript density bin maps to the upper end of the palette (i.e., Inferno and Viridis=yellow), with a linear distribution in between. The Density map scale threshold slider allows you to choose a different mapping range (units = transcript count per µm2), which can help to visualize bins that are at or beyond the limits of the density range.
When displayed as points, checked gene icons will be visible in the viewing area and unchecked genes will be invisible. Low quality transcripts (Q-Score < 20) are filtered out by default. Switch the toggle to display them as gray circles (note: these are not the same as "Ungrouped" gene gray icon points).
As you explore the data and zoom in/out, Xenium Explorer will display the number of transcripts in the viewing area per selected gene next to the gene name.
The individual transcript or gene group icon and color can be changed (depending on whether point style is "Icons" or "Circles"). You can reset to the original (prior to creating grouped genes) point color and icon by selecting Reset to default. Transcripts are plotted by default as small points, with the ability to customize point size and type to enable better visualization of transcript distribution across the sample.
Xenium Explorer v3.0 introduces scaled transcript visualization, which clusters transcripts together based on gene identity and spatial localization. This algorithm enables Xenium Explorer to maintain the performance that is essential for exploring in situ data, while visualizing the spatial transcript distribution for both highly and lowly expressed genes, at any zoom level.
Scaled transcript visualization requires clustered transcripts from the transcripts.zarr.zip file pre-computed by Xenium Onboard Analysis v3.0 and later. It can only be viewed in Xenium Explorer v3.0 and later. For data generated with previous XOA versions, Xenium Ranger v3.0 and later enables the generation of transcripts.zarr.zip in the format required for scaled transcript visualization.
At each zoom level, for each gene, neighboring transcripts are grouped into a single, larger point if they are within an initial 16 pixel radius of each other. This radius doubles in size as you zoom out. The point size is calculated as the square root of the number of transcripts in each aggregate point multiplied by a scaling factor. Each point is labeled with the number of transcripts it represents.
-
"Scaled transcripts view enabled" is displayed in the bottom left corner of the window to indicate that transcript points are scaled. Points that represent groups of transcripts will not show transcript-specific information such as Q-Score, ID, or location.
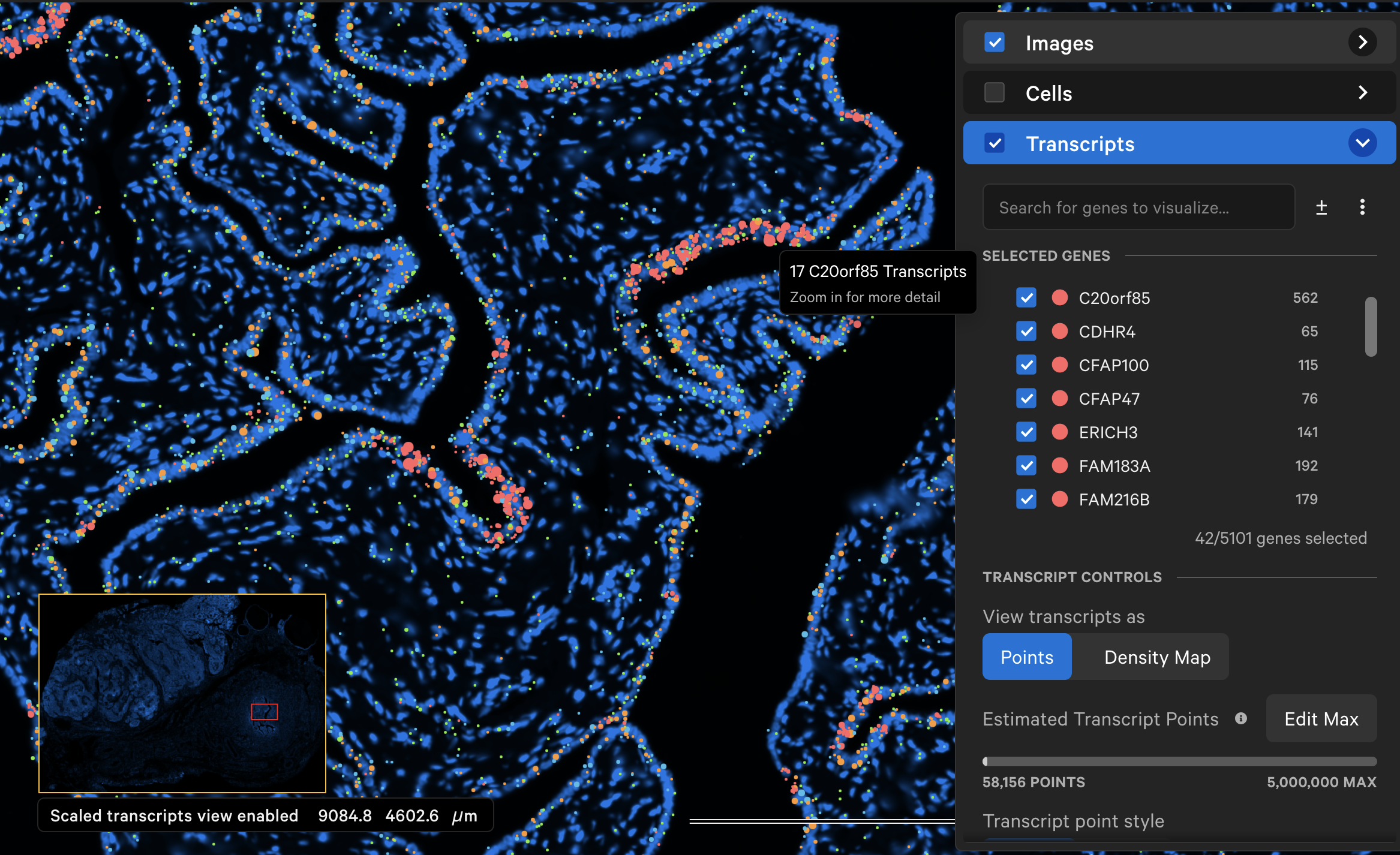
-
The exact zoom level at which the scaled view turns off and all transcripts are plotted can vary across datasets (track zoom level in Image options). When zoomed all the way in, individual transcript points are shown.

First, decide which genes you want to create groups for. The cell_feature_matrix/features.tsv.gz file in the output directory contains the full list of pre-designed panel genes, as well as any custom add-on genes. For each feature, the Ensembl ID and gene name are stored in the first and second column of the features.tsv.gz file, respectively. The third column identifies the feature type (Gene Expression). You can copy/paste a list of Gene Expression genes from the second column of this file. Xenium Explorer does not use the negative control rows (i.e., Negative Control Probe, Negative Control Codeword, Unassigned Codeword); they are used for calibration during the on-instrument analysis.
# to view the file
gzip -cd features.tsv.gz | less
# to uncompress the file
gunzip features.tsv.gz
Example:
ENSG00000166535 A2ML1 Gene Expression
ENSG00000127837 AAMP Gene Expression
ENSG00000131043 AAR2 Gene Expression
ENSG00000266967 AARSD1 Gene Expression
ENSG00000183044 ABAT Gene Expression
ENSG00000165029 ABCA1 Gene Expression
ENSG00000167972 ABCA3 Gene Expression
[...]
Next, create a gene group comma-separated value (CSV) file in a text editor, such as Microsoft Excel. The format of the CSV file is as follows:
- The first row must have two column headers in this order with these exact names: "gene" and "group".
- The first column is a list of gene names.
- The second column is a list of group labels. The CSV file may contain an arbitrary number of columns for other group labels.
Here is an example from the Xenium Prime 5K FFPE human ovarian cancer dataset (download custom gene group file here):
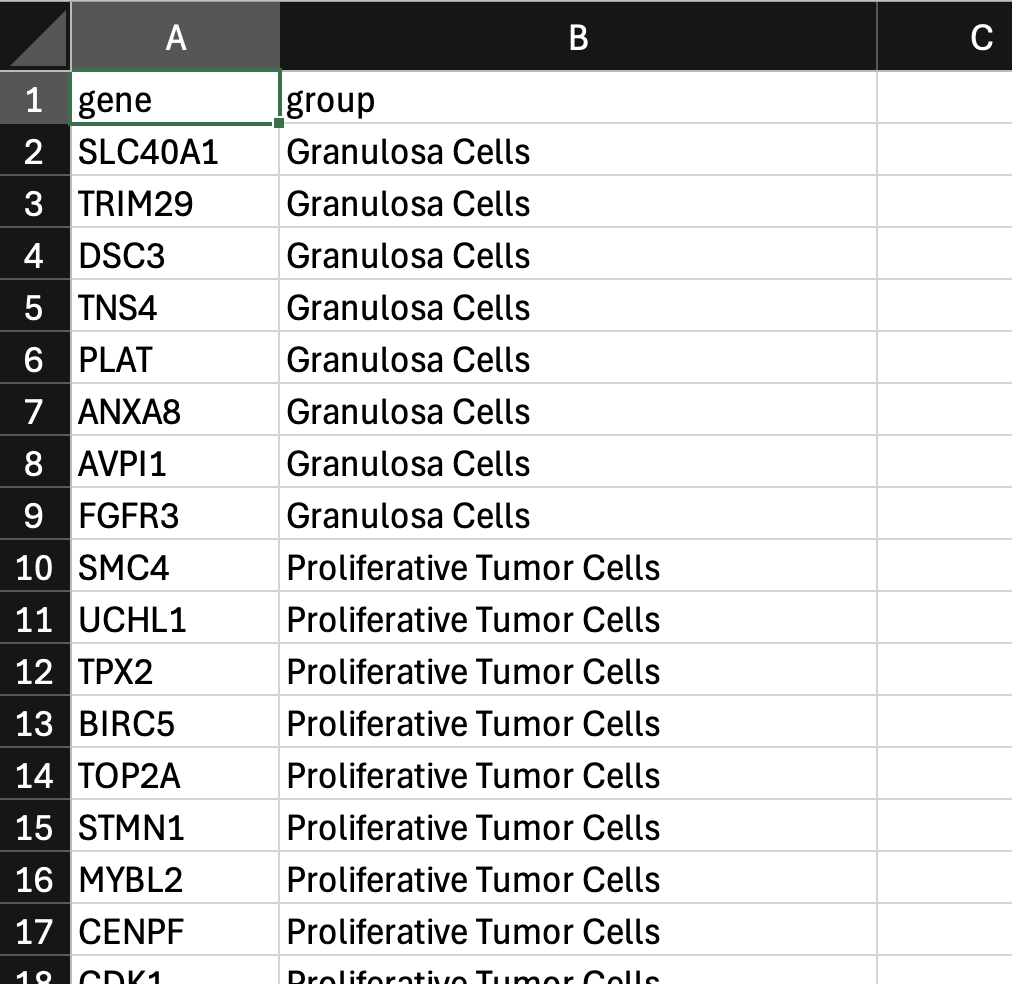
Next, click the three vertical dots and select Create gene groups. Click Upload Gene Group CSV and choose your CSV file.
The gene list will now display the group names with dropdown menus. Grouped genes are automatically assigned the same point color. If panel genes were not included in the CSV file, they will be automatically grouped into an "Ungrouped" category. Ungrouped genes have a gray circle icon by default.
If the CSV file contains multiple group name columns, all group names will be listed alphabetically. Individual genes may be assigned to multiple groups and will be listed under each group name.
The imported gene list and associated transcript icon color and shape changes can be saved with the Saved Current Views option.
In Xenium Explorer v3.2, the localization plots gallery button is now located on the top menu bar. Per-gene localization plots are generated for all Xenium datasets, while per-codeword localization plots are available for custom and boosted genes in Xenium Prime 5K datasets
The per-gene localization feature is useful for assessing the spatial location of genes in the entire sample area. It displays spatial density map plots of total transcript counts (Q-Score ≥ 20) per gene and for all selected genes (combined counts if more than one gene is selected).
For Xenium Prime 5K datasets, gene detection can be split over multiple codewords (gene splitting). Xenium Explorer v3.2 adds a per-codeword localization gallery for custom and boosted genes.
It displays spatial density maps of total transcript counts (Q-Score ≥ 20) per codeword for each custom or boosted gene.
If present, boosted genes will be assigned their own category ("Boosted Genes") in the Transcripts search list. The per-codeword localization gallery enables visualization of transcript counts and spatial distributions per codeword, offering greater insight into the gene expression patterns.