Open Xenium Explorer by double-clicking the application icon.
There are a few options to open datasets in Xenium Explorer (see Xenium Explorer Input Files). You can drag and drop or click Open New File to select the .xenium file. There is also a dropdown menu to either Select file (same as Open New File) or Open file from path. If you open a file from path, enter the path to the location where the .xenium file is stored on your computer or network drive.
Starting in Xenium Explorer v4.0, you can also load XOA files from the AWS cloud. Instructions are provided here.
The Xenium Onboard Analysis output files must be in the same directory path as this file (unless you have modified the file paths).
Once you have opened a dataset, the Xenium Explorer home page displays options to open or clear recent files. Metadata are shown on the recent file tiles. Up to 100 recent datasets will be displayed in Xenium Explorer v4.0 and later.
When you close a dataset, the view settings will be saved automatically as an unnamed saved view (similar to explicitly saving current views). If you open a dataset from the Recent Files menu, Xenium Explorer will restore your last view. However, if you open the same dataset from outside the Recent Files menu (drag and drop, double-click .xenium file, or Open New File), Xenium Explorer will open with the default initial view (grayscale DAPI image) and the unnamed saved view will be lost. The best way to ensure you can reopen the view state again later is to save your current view before closing Xenium Explorer.
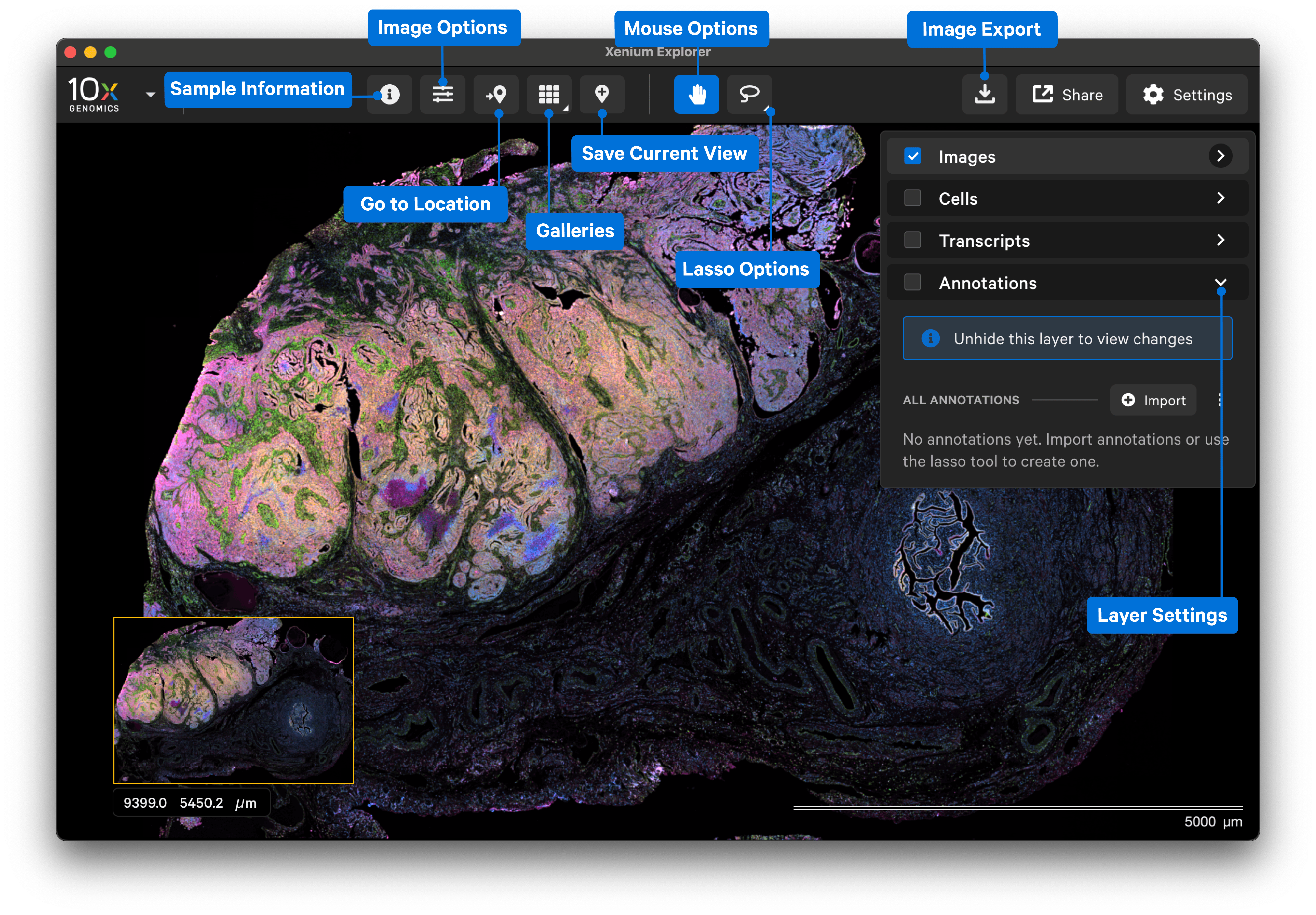
After the dataset loads, you should see images of the stained cells and/or nuclei. The following image shows the key components of the Xenium Explorer interface. In this Getting Started tutorial, we provide a brief overview of the image, cell, transcript, and lasso features. The Save Current View and Share features are described in the Saving and Sharing Results tutorial.
The Sample Information pop-up window displays metadata about the sample run, analysis, and panel. The Run and Panel sections show information entered on the Xenium Analyzer instrument during the run set up; they are stored in the experiment.xenium manifest file. From the Analysis section, you can open the analysis summary HTML file in another Xenium Explorer window.
This window also displays the "Panel Design ID" for Xenium Gene Expression panels and if used, Xenium Ranger metadata.
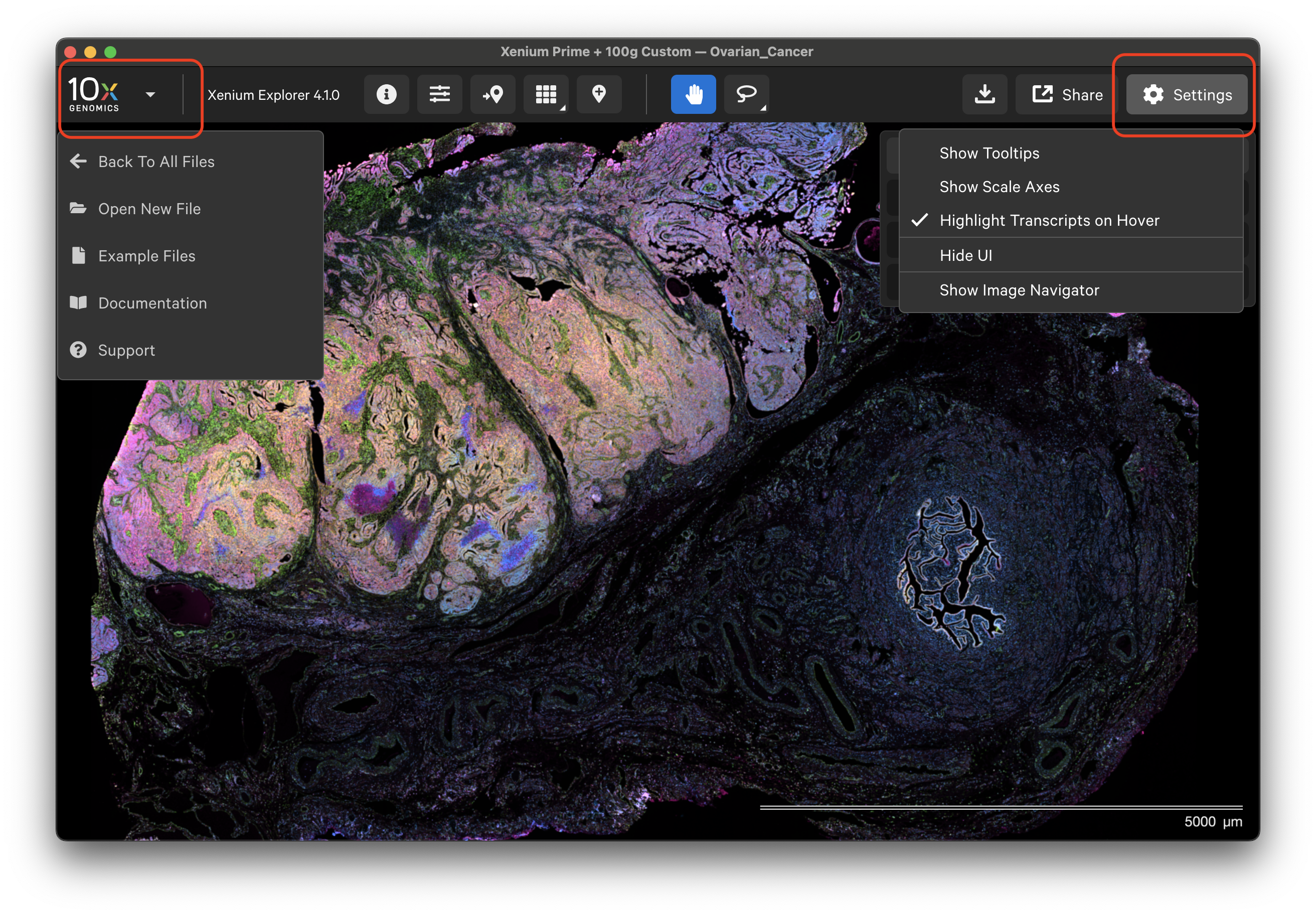
Click the 10x Genomics icon in the top left-hand corner to return to the home screen, open a new dataset, or find support documentation.
The Xenium Explorer interface settings are controlled in the Settings menu and with keyboard shortcuts. The Settings menu allows you to show or hide transcript and cell tooltips, image scale axes, the user interface (UI) itself, and the image navigator in the bottom left-hand corner.
Keyboard shortcuts:
| Key | Alternative | Description |
|---|---|---|
| Tab | Settings button | Show/hide UI |
| = (equals key) | Scroll with mouse | Zoom in |
| - (hyphen key) | Scroll with mouse | Zoom out |
| L | Lasso button | Draw a freehand shape with lasso selection tool |
| R | Lasso button | Draw a rectangle with selection tool |
| P | Pan button | Move the image up and down, or left and right |
Click the download button in the upper right corner to select an image export option. Exported images are saved in PNG format for both options.
Use the Settings options to show or hide the axes ("Show Scale Axes") and the picture-in-picture image ("Show Image Navigator"). The Xenium Explorer UI menus are not included in the exported image.
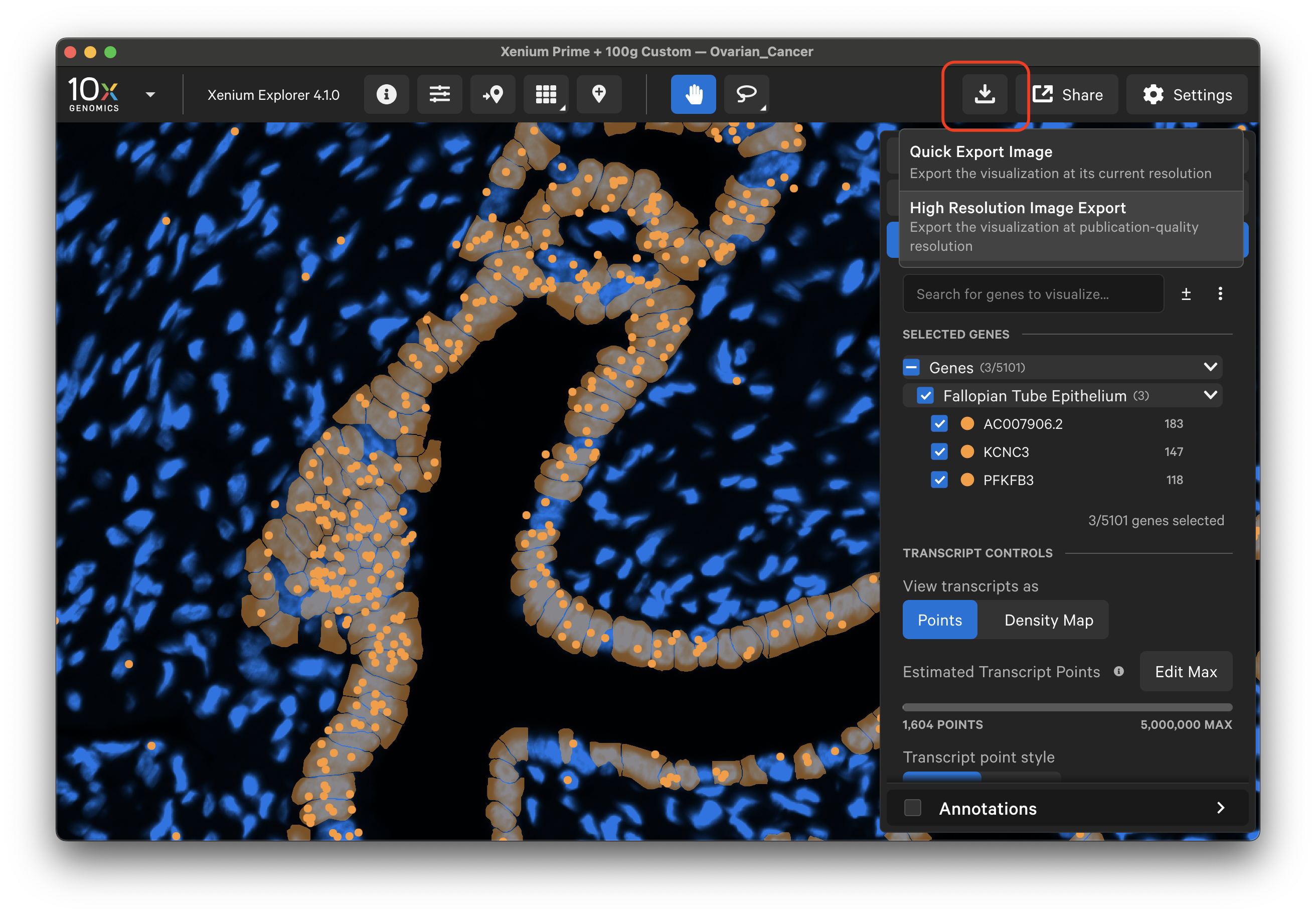
Quick Export Image
Select "Quick Export Image" to export screenshots of image, cell, and transcript information displayed in the Xenium Explorer viewport at their displayed resolution.
High Resolution Image Export
Starting in Xenium Explorer v3.2, select "High Resolution Image Export". The exported image will match the aspect ratio of the current viewport and include all visible elements of the visualization, without the user interface.
Xenium Explorer generates the high resolution image by stitching smaller image tiles together, which will appear to flash on-screen as they load. The exported image will include more detail than is currently visible.
- When zoomed in (Image options zoom level >= 0.25), the app zooms in further to the maximum level (8) to capture the image tiles. If points are selected, scaled view is turned off and all transcripts are plotted.
- When zoomed out (Image options zoom level < 0.25), the app zooms in further 32x the level of the current image to capture the image tiles. If points are selected, scaled view is turned on and plotted at 32x the level of the current image as well.
Transcript points can appear smaller in the exported file than in the app viewport, so we recommend increasing the "Transcript point size scale" to ensure points are visible in the exported file. Depending on the region size, exporting may take a few seconds to several minutes.
The Images, Cells, Transcripts, and Annotations layers allow you to overlay multiple pieces of information in the explorer viewing area.
The Images layer shows settings to adjust the 3D image stack of the nuclei-stained (DAPI) cells and autofocus stain image(s). Image import and alignment workflows are also available in this layer. The Image layer is automatically checked when Xenium Explorer is opened. Click the links below to learn more.
The Cells layer displays nuclei and cell borders based on cell segmentation results. Check the box next to the layer name to view. Click the link below to learn more.
The Transcript layer displays transcript count data. Check the box next to the layer name to view. Click the link below to learn more.
The Annotations layer displays polygons for selected regions of interest that are both created in Xenium Explorer or imported from third-party tools. Check the box next to the layer name to view.
Click the link below to learn more.
To annotate Regions of Interest (ROIs) in Xenium Explorer, there are two lasso shape options - rectangle and freehand lasso. Here's an example using the freehand lasso tool to count genes per cell a region with CFTR- tumor cells (see pancreatic cancer explorer page):
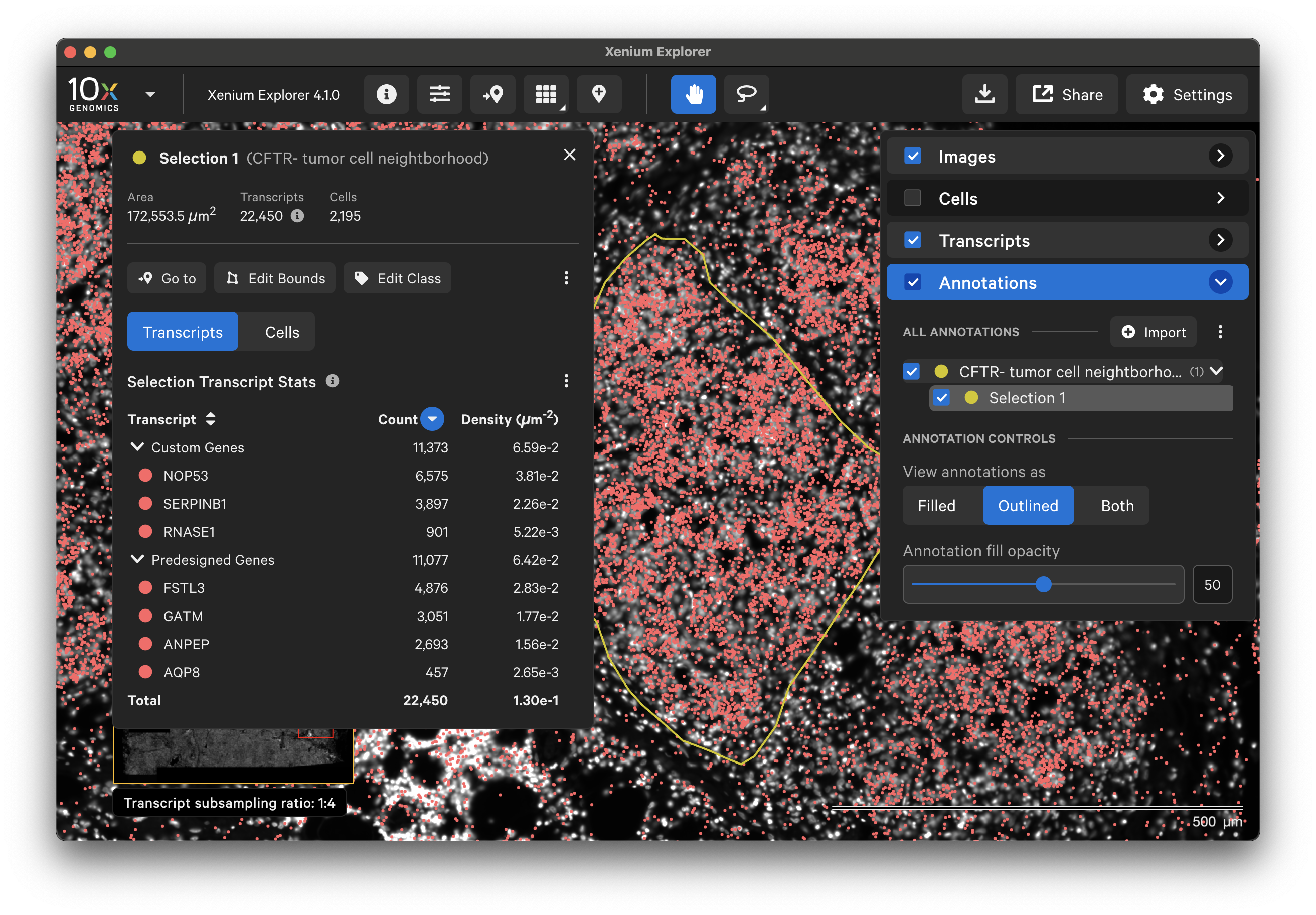
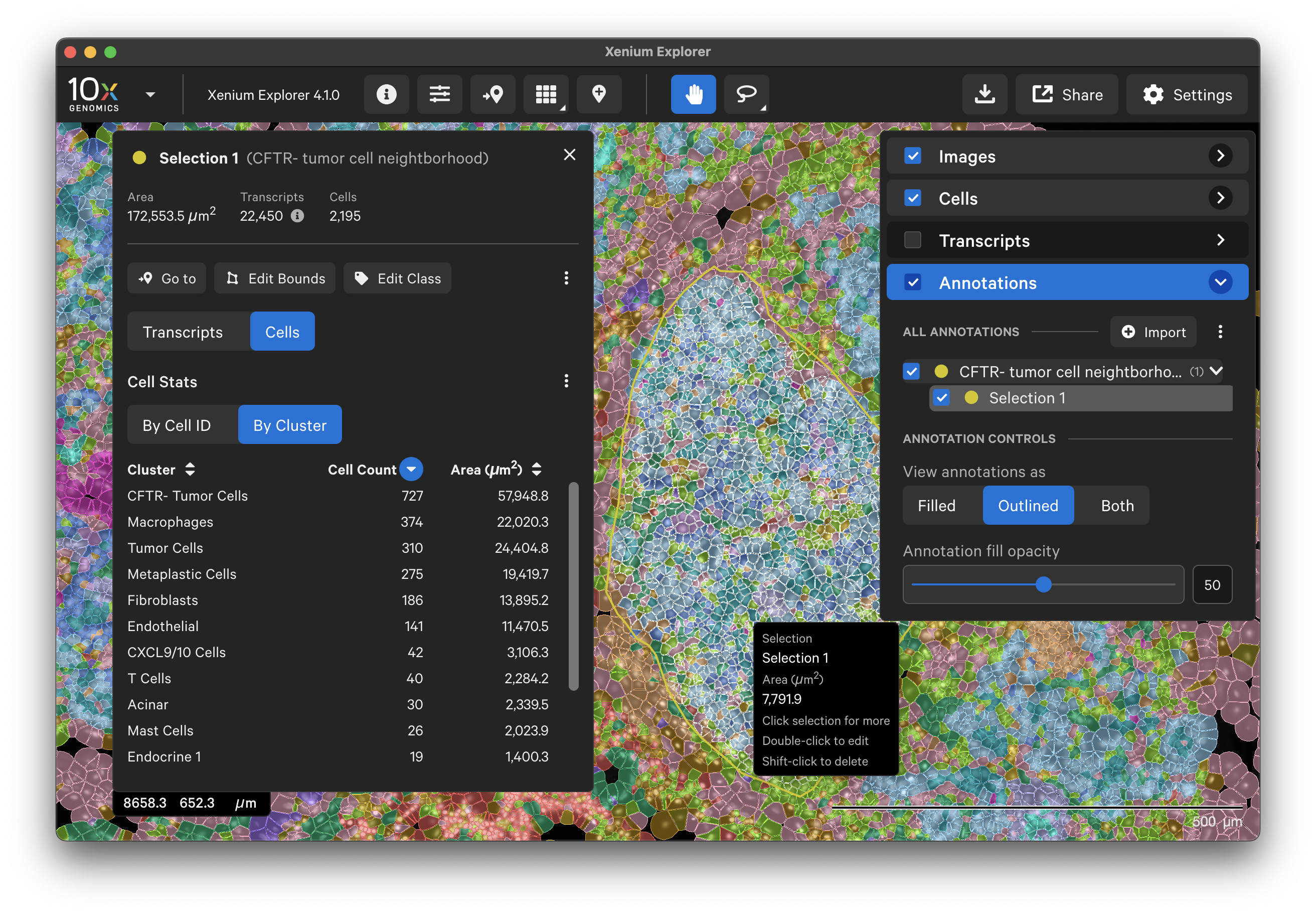
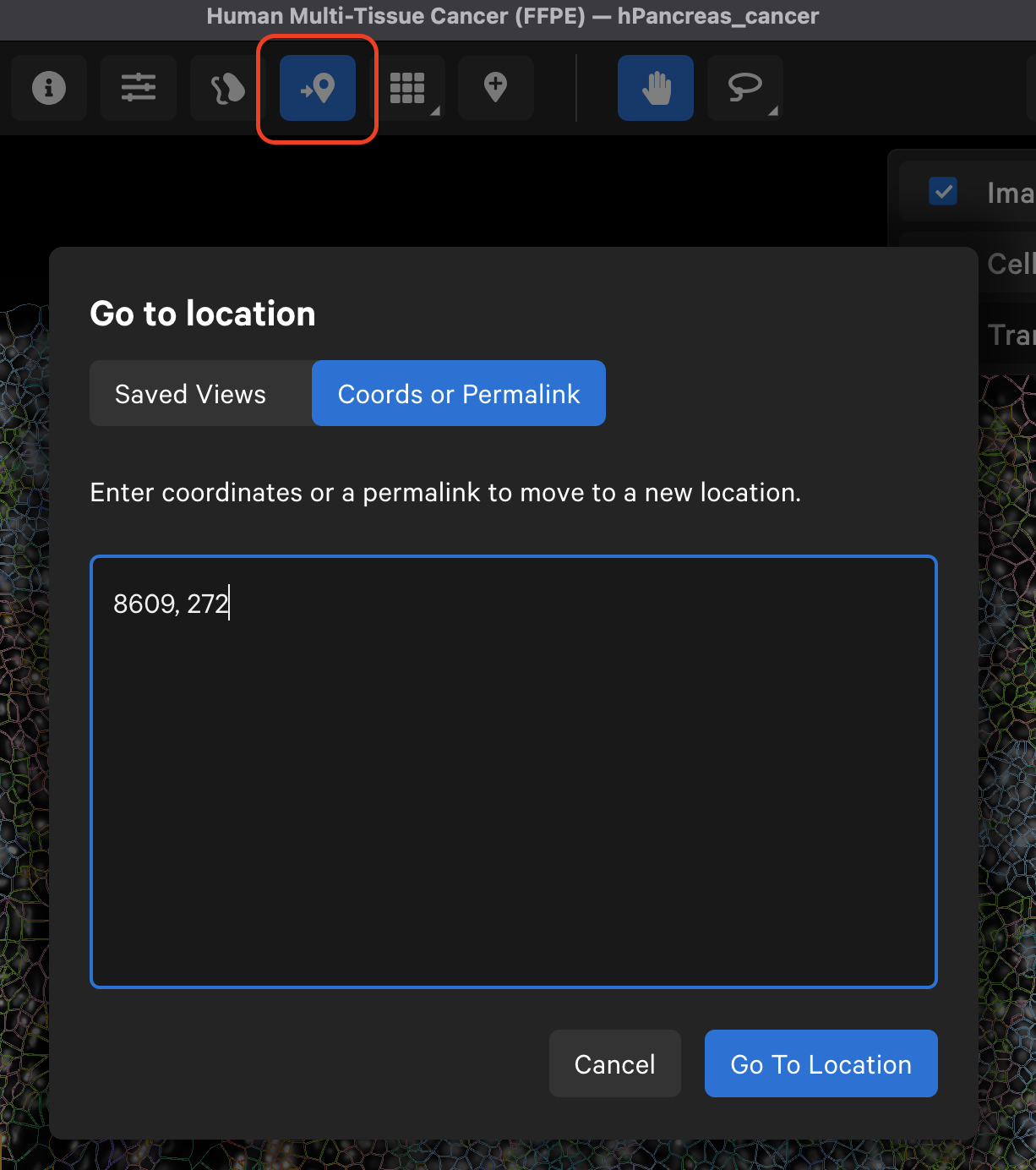
The Go to location button enables you to jump to a specific X-Y coordinate location in the image, open a saved view, or transfer a permalink view from another session. X-Y coordinate values are shown in the lower left-hand corner of the viewing area in µm. The X-Y format must be comma-separated (i.e., 8609, 272). Lists of coordinates from selected Regions of Interest or cells should be pasted with the Lasso button’s Paste coordinates feature (see Multiple Selections tutorial).
The experiment.xenium manifest file contains metadata read in by Xenium Explorer about the experiment, including the paths to the input files listed above. The file names in the images and xenium_explorer_files sections use the default output file names from Xenium Onboard Analysis.
In order to view and interact with custom analysis results in Xenium Explorer, you can edit these sections of the manifest file. Because the experiment.xenium file is a text file in JSON format, changes can be made in any text editor. The file paths can be absolute or relative. However, relative paths should not use a tilde (~) for home directory path expansion.
Xenium Explorer v3.0 - 3.1 do not support certain transcript and cell functions due to performance limits for extremely large Xenium datasets with > 200 million unique cell by transcript features (sparse matrix). These functions are restored in Xenium Explorer v3.2 and later for XOA v3.1 and later datasets.
- For cells in selected regions of interest, transcript information is not available.
- For individually selected cells, transcript and cell statistic information are derived from cell polygons instead of the segmentation mask.
- Cell tooltips will not display transcript counts
- Cells cannot be viewed by transcript density.
This example Unix command can be used to check the size of your dataset's cell-feature matrix Zarr file (be sure to edit the path to your matrix file):
# Extract matrix file to pipe
# Extract shape of the matrix
unzip -p cell_feature_matrix.zarr.zip cell_features/data/.zarray | grep shape --after=1
# Example output: this cell-feature matrix has ~158 million cell x transcript features
# "shape": [
# 157829388